
A Risk-Based Approach To Filter Integrity Testing Annex 1 Requirements For Biologics DS
By BioPhorum

The new revision of EudraLex Annex 1 aims to provide guidance for the manufacture of sterile drug products (DP). However, it also suggests that some of its principles and guidance may be used to support the manufacture of low bioburden biologicals (active pharmaceutical ingredients/drug substances (DS)). As in the manufacture of sterile products, manufacturing low bioburden biologicals is subject to special requirements to mitigate the risks of viable and non-viable contaminants. Annex 1 includes a comprehensive list of criteria for the design, installation, operation, and testing of sterilizing-grade filters. The list includes an expectation that the filtration system be designed for pre- and post-use filter integrity testing (FIT).
The question is, how do the principles of FIT described in Annex 1 apply to DS manufacturing?
For readers focused on DS manufacturing facilities, the intentional ambiguity in Annex 1 can raise questions about where current procedures and policies stand regarding FIT and regulatory compliance for filters that are not routinely integrity tested. While the new version of Annex 1 is relatively explicit about FIT requirements for DP facilities, there is no clear guidance from health authorities or industry associations on FIT requirements in DS manufacturing.
This article aims to help drive alignment across industry and identify best practices for FIT.
The consensus of available guidance suggests using an application-dependent, risk-based approach. As such, a systematic risk assessment tool designed to mitigate risks to product, process, patient, and business was used to evaluate multiple categories of sterile and bioburden reduction filtration operations used in a common DS manufacturing facility. The purpose was to illustrate where strong risk mitigation controls are recommended and where FIT provided little or no value (and is therefore not recommended). It also includes a model decision tree evaluation process to determine which sterilizing-grade filters should undergo integrity testing when used in typical DS manufacturing schemes.
Background On FIT And Annex 1 Requirements
To set a solid foundation for this analysis, key definitions and regulatory requirements for FIT should be established. Starting with a definition, the Annex 1 glossary states that FIT is: “A test to confirm that … [filters] (product, gas or HVAC filter) retain their retentive properties and have not been damaged during handling, installation or processing.” In simple terms, it is a test to prove the filter works (or worked) as intended.
The FIT can be performed pre- or post-use and in-place or out-of-place (i.e., remotely). The guidance in Annex 1 is:
“8.82 The filtration system should be designed to: … permit in-place integrity testing of the … final sterilising grade filter, preferably as a closed system, both prior to, and following filtration as necessary. In-place integrity testing methods should be selected to avoid any adverse impact on the quality of the product.”
This statement is critical and endorsed by the authors of this article. For DP manufacturing, FIT is an integral part of the overall contamination control strategy and validation of sterile filling operations because it is not physically possible to test every dose that may reach a patient. Therefore, the FIT of critical filters in sterile manufacturing is essential.
Application Of Annex 1 Guidance For DS
Turning to the new scope excerpt in Annex 1, it is suggested that the principles required for DP manufacturing may also be applied to active pharmaceutical ingredients:
“The intent of the Annex is to provide guidance for the manufacture of sterile products. However, some of the principles and guidance, such as contamination control strategy … may be used to support the manufacture of other products that are not intended to be sterile such as … low bioburden biological intermediates.”
This may lead some DS manufacturers to believe the FIT requirements outlined in Annex 1 should be applied to low bioburden DS operations, but to what extent? To be conservative, one might consider the lowest risk approach to be 100% testing of all sterilizing filters, both pre-use post-sterilization integrity test and post-use. However, considering the principles of continuous improvement and following ICH Q10 guidance for quality risk management, that approach may not be value-added or even necessary.
To reach a sound conclusion, a risk-based approach was used to determine the value of FIT for DS manufacturing. A failure modes effect analysis (FMEA) was used; however, other risk-assessment platforms may be appropriate as deemed by the end user.
Scope Of Analysis
This evaluation aimed to assess the guidance in Annex 1 for gaps when applied to a low bioburden biologics facility; therefore, only DS manufacturing processes were included in the scope. Only process filters were evaluated; utility gas distribution filters were out of scope. Furthermore, the evaluation focused on sterilizing grade filters, so other specialty filters (e.g., viral retentive filters, ultrafiltration/diafiltration membranes, depth filters) were also out of scope.
The scope of the analysis was based on a typical monoclonal antibody manufacturing facility’s process flow and equipment plan. A process plan depicting all sterilizing-grade filter categories in the bioprocess was generated to help ensure all filters were considered and enabled the grouping of filter classes (where appropriate), by area, operation, and type to generate a comprehensive list of unique filters.
Some Of The Assumptions And Criteria Used In The Analysis
- Risk tolerance is inherently dependent on the end user and their unique factors. Impact assessments may be based on industry consensus, but every case or application needs to be assessed individually.
- The analysis focused on low bioburden processing. DS manufacturing is distinct from sterile DP manufacturing in that failure of a sterile filtration is often detectable by other means (e.g., bioreactor contamination or bioburden testing) before product is administered to a patient.
- Patient and business risks should be considered in an overall risk assessment. Although contamination of a DS may result in a low risk to the patient due to detectability, the loss of a batch could represent a significant cost impact and interruption of supply to the patient.
Execution Of The FIT Risk Assessment
For an FMEA-type risk assessment, the failure mode describes the type of failure and how it could occur (e.g., a filter is non-integral due to damage from SIP), and the effect is the outcome of the failure (e.g., filtered material may not meet sterile filter claim). The risk of the failure mode and its effect is then quantified by the severity of the effect, the likelihood of its occurrence, and the detectability of that failure:
- Severity — The impact or consequence of the effect of the failure.
- Likelihood — The probability that the failure mode occurs and causes the effect.
- Detectability — The ability to identify the failure mode.
To ensure consistent results and a systematic analysis, preestablished scoring criteria for each element were defined and used throughout the risk assessment; these are summarized in Table 1.
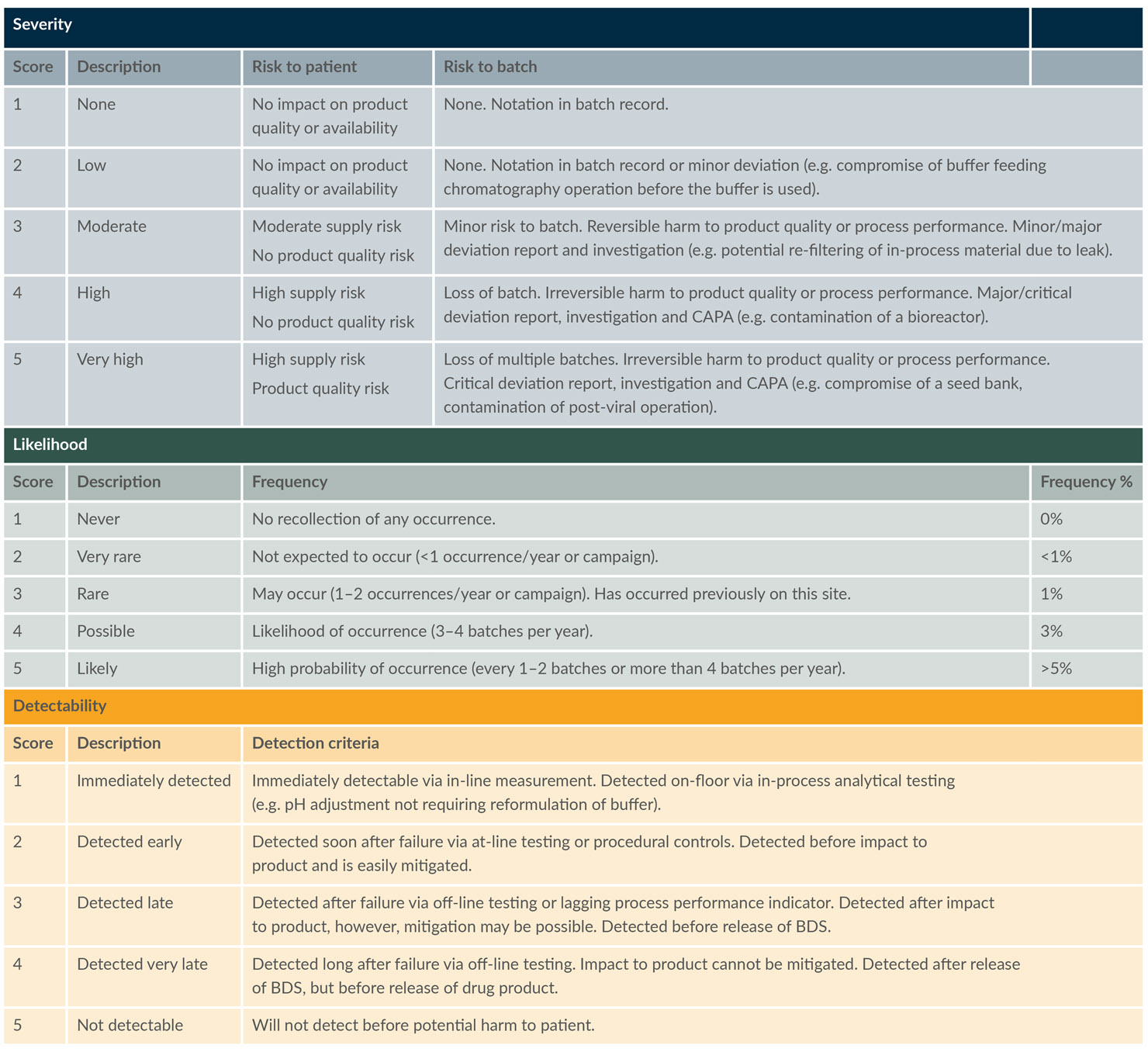
Table 1: Scoring criteria used for FMEA risk assessment. (click to enlarge)
Key: BDS – Bulk drug substance, CAPA – Corrective and preventive action
A database tool can be used to execute the FMEA risk assessment. The authors scored the likelihood of failure of a sterilizing-grade filter and its potential impact on the step, overall process, and patient. The ability to detect the failure with and without FIT also was evaluated. The database tool then compiled the data and used the preestablished scoring criteria in Table 1 to arrive at the risk index (RI) and the risk priority number (RPN) for each potential failure mode.
Risk index = Severity x likelihood
Risk priority number = Risk index x detectability
The FMEA provided an output that helped determine where FIT could provide value to mitigate risk to the process. Using the grids in Figures 1 and 2 below, the RI and RPN were categorized as Low, Medium, or High.
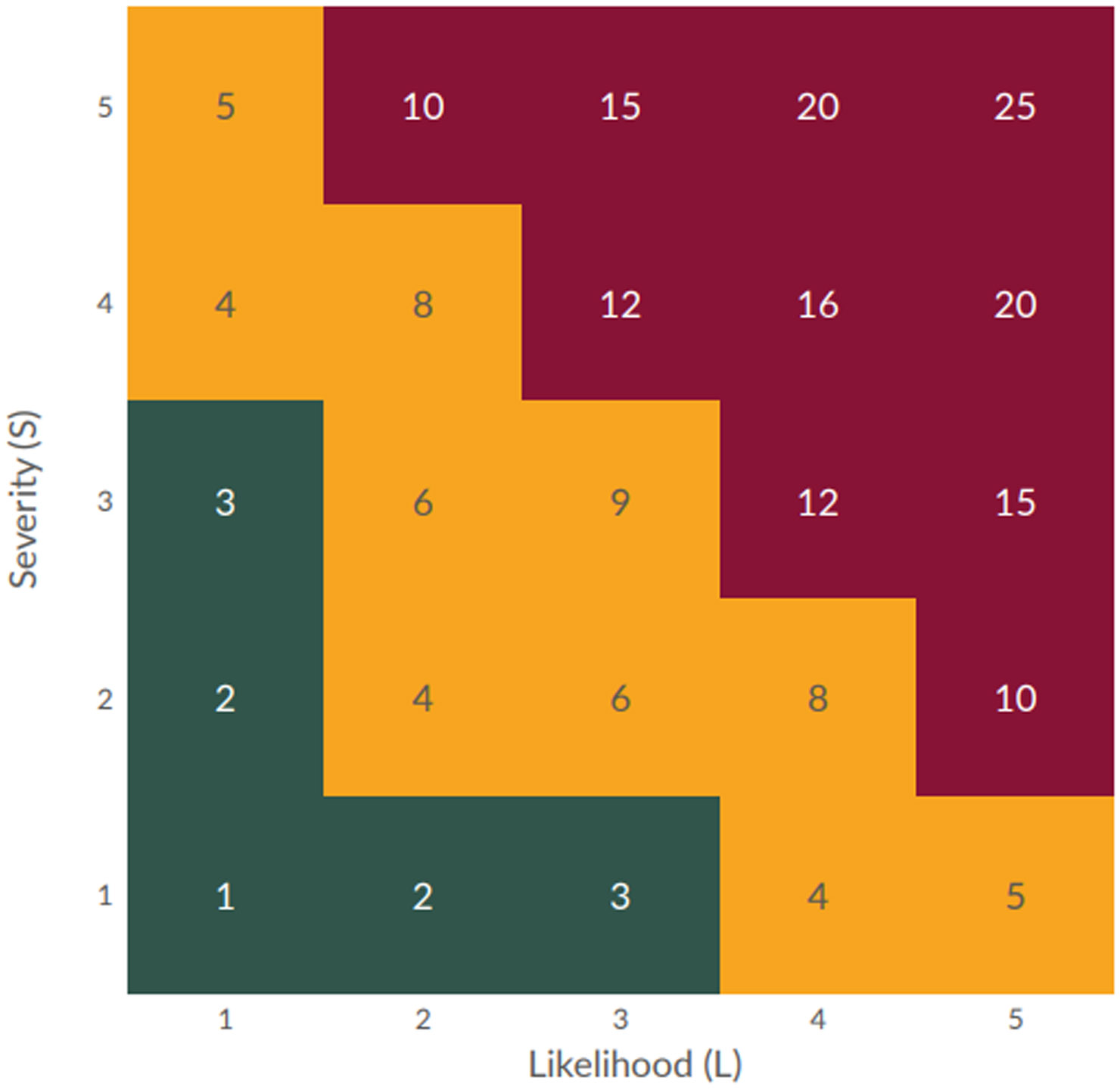
Figure 1: Risk index grid (click to enlarge)
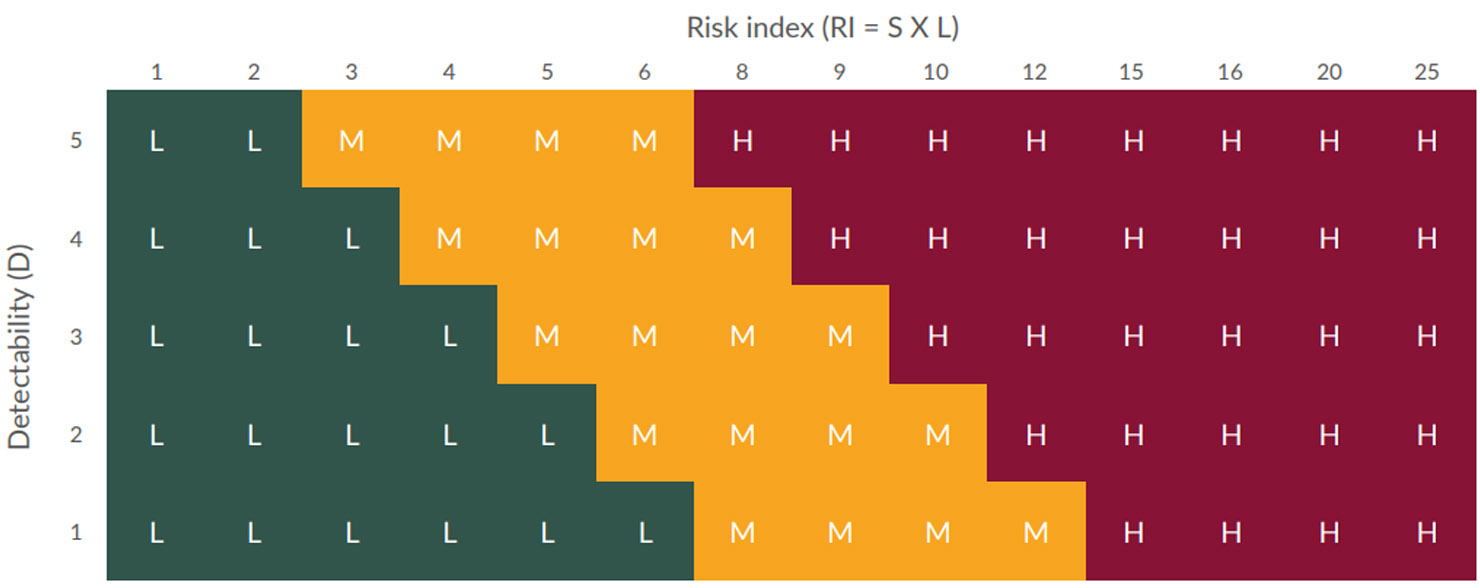
Figure 2: Risk priority number grid (click to enlarge)
- Where RPN was categorized as Low (without FIT), FIT is not recommended.
- Where RPN was categorized as Medium (without FIT) and applying FIT reduced the risk to Low, FIT may provide value and is suggested.
- Where RPN was categorized as High (without FIT) and applying FIT reduced the risk to a Medium or Low, FIT does provide value and is recommended.
Table 2 shows assessments for two scenarios where sterilizing grade filters are used: a vent filter on a buffer preparation vessel (scenario 1) and a bioburden reduction filter used for a chromatography eluate (scenario 2, where the eluate is collected and held before processing. If this is a continuous process, the likelihood and severity may both be lower). Each failure mode includes multiple effects, which are shown in separate rows to distinguish the potential outcomes.
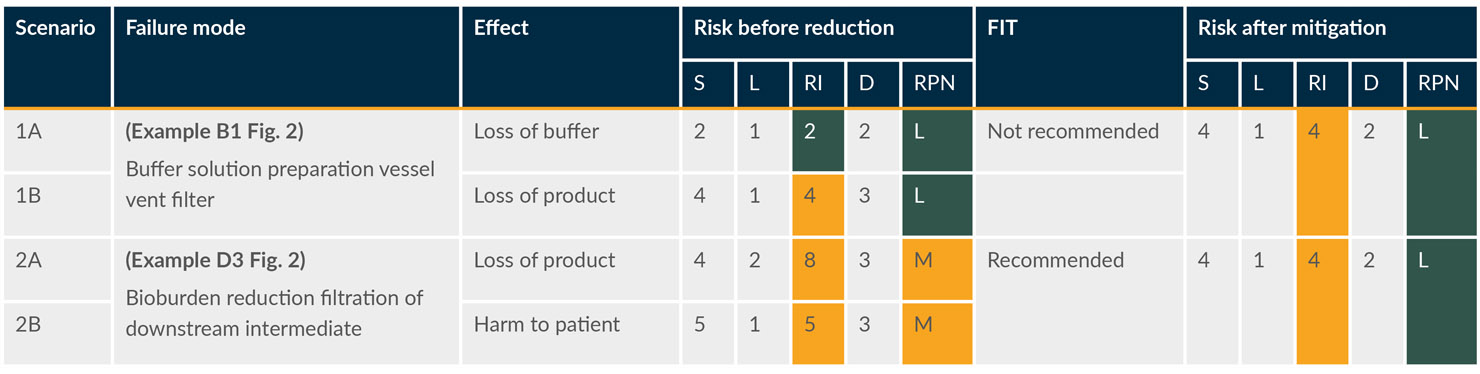
Table 2: Excerpt of the risk assessment table. (click to enlarge)
Key: D – Detectability, L – Likelihood, S – Severity.
FIT Risk Assessment Decision Tree
The FIT risk assessment was executed in this way for all filters (involving approximately 100 unique situations). On completion, it was clear that the scores and FIT recommendations depended on a short list of questions. This realization enabled more consistent logic for broader alignment across all unit operations.
This led to a simplified risk-based approach using a decision tree model. The newly designed FIT decision tree is a highly efficient tool that simplifies the overall risk-assessment process and promotes enhanced communication of major conclusions in an easy-to-follow and easy-to-apply format.
These questions were used to create the decision tree:
- Is the solution used in low bioburden or sterile/aseptic operations?
- Is the solution being filtered directly into the product?
- Is the solution bactericidal, bacteriostatic, or growth-promoting?
- Is the filtrate being stored or is the filtrate being forward-processed immediately?
- Is there a known risk in the filter installation procedure?
- .Is the filtrate an excipient, final formulation buffer, or final bulk DS?
The resultant decision tree (Figure 3) is a simple and systematic method to determine if a filter integrity test is recommended and aligns with the overall FMEA performed in this study.
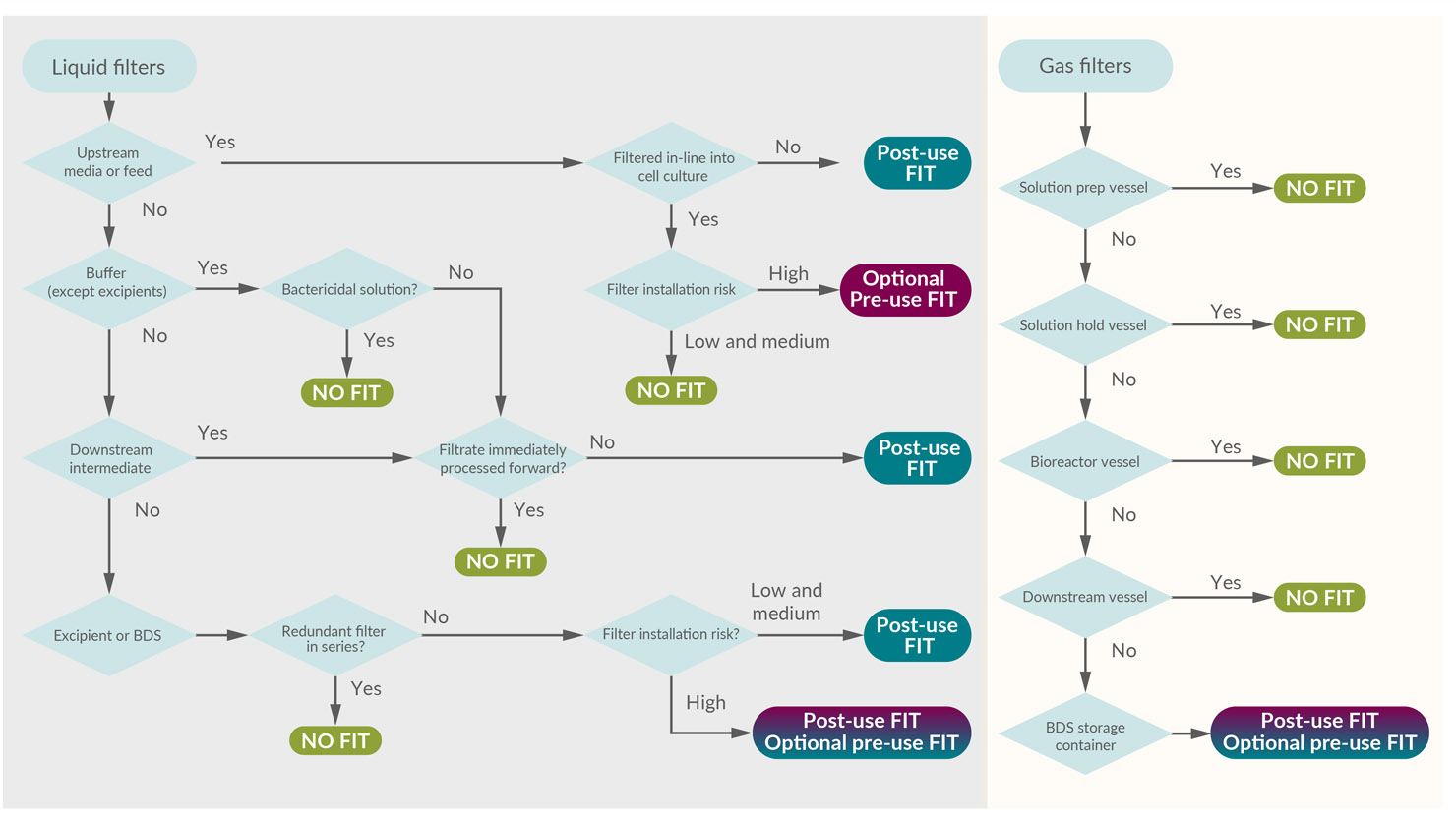
Figure 3: Risk-based decision tree to determine the value of a filter integrity test. (click to enlarge)
Key: BDS – Bulk drug substance.
To illustrate alignment with the risk assessment, the example of the downstream intermediate bioburden reduction filter will again be evaluated in the decision tree. The FMEA approach results in a FIT recommendation, as shown in Table 2. Following this through, the FIT decision tree yields the same recommendation. The path is highlighted in Figure 4.
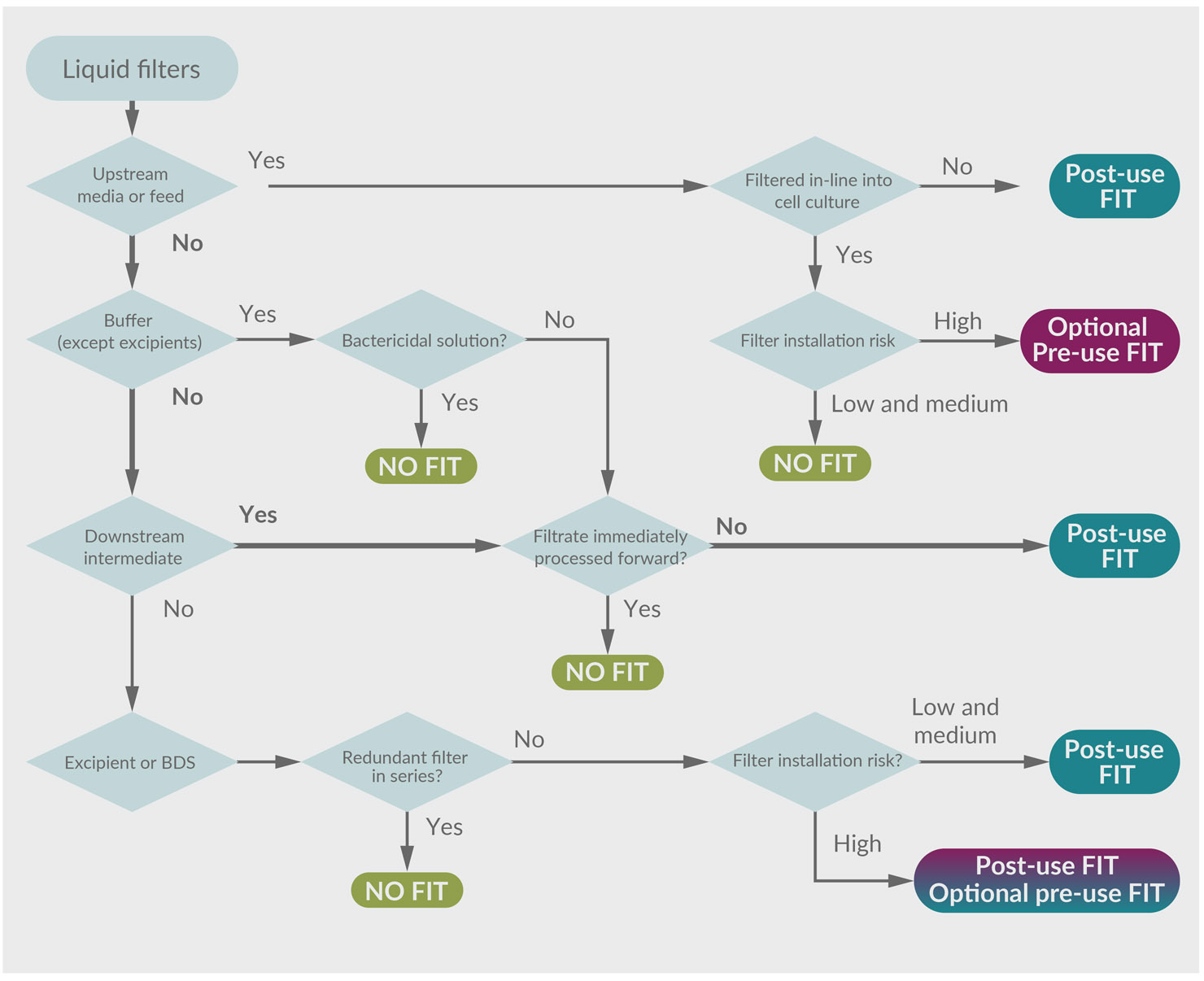
Figure 4: FIT decision tree with highlighted path. (click to enlarge)
Key: BDS – Bulk drug substance.
In this example, the hold step provides an opportunity for microbial proliferation that could impact product quality. Performing a FIT before the release of the intermediate to subsequent processing could detect that risk. The ability to confirm process integrity is consistent with a robust contamination control strategy. Interestingly, had the process intermediate been processed immediately without a hold, the FIT would serve little value and would not be recommended (except maybe for forensic purposes).
The risk-based FIT decision tree is intended to serve as a model and may not represent every application. DS manufacturers should interpret and apply the model appropriately, considering their unique and specific risk factors.
Conclusions
Industry practices for FIT deployment are not harmonized with the variety of approaches used. Many DS manufacturing companies are performing FITs that may provide limited or no value as a risk mitigation measure in a contamination control strategy.
The recent revision of Annex 1 provides guidance and expectations around FIT with a special focus on pre-use and post-use testing. However, applying this guidance universally to low bioburden DS manufacturing (Annex 2) may be excessive. A risk-based approach should be followed for determining when and where to perform FIT. Based on this article, a decision tree model could be adapted to simplify the risk assessment. Each company may develop its own short list of questions and/or a similar decision tree to aid the consistent and accurate evaluation of the need for FIT performance.
By considering the true value and mitigation provided by each FIT performed, DS manufacturing sites can focus limited resources on those areas best positioned to improve process execution, product quality, and patient safety outcomes.
This article summarizes some of the main points from a recent BioPhorum publication on this topic. To read more, including an example of a completed FMEA risk assessment, check out the full paper, A risk-based approach to filter integrity testing requirements for biologics drug substance manufacturers.