
Aligning Sterile Filtration With EU GMP Guide Annex 1 Standards
By Raimund Brett, GMP Compliance Adviser

Sterile filtration is a crucial process step in the manufacture of sterile pharmaceutical products from heat-sensitive starting materials. In contrast to other sterilization methods, the microorganisms are not inactivated or killed, but removed. Sterile filtration, which is used to remove particles — both living (microorganisms) and non-living — from liquids and gases, is one of the most important steps in the aseptic processing of sterile medicinal products including heat-labile materials.
Important aspects of sterile filtration are the qualification of the filter, the validation of the filtration process, and the execution of filter integrity tests. Sterile filters must be sterilized before use. After filling a batch, at the latest after one working day, the sterile filter should be disposed of. For small batches, reuse as a pre-filter can be considered for cost reasons but requires appropriate validation.
Suitable pre-filters and sterile filters may be used in the process to control and maintain a low product bioburden. This serves to ensure the success of the final sterile filtration. Filters with a nominal pore size of 0.2 µm to 0.22 µm are used as sterile filters. The filter must be compatible with the product and correspond to the description in the marketing authorization (EU GMP Guide Annex 1, paragraph 8.79).
The design of the filtration system (filter and connections) should be established to fulfill the following requirements (EU GMP Guide Annex 1, paragraph 8.82):
- Allow operation under the validated process parameters (e.g., temperature, viscosity, pressure, etc.)
- Maintain the sterility of the filtrate
- Minimize the number of aseptic connections between the sterilizing filter and final filling of the product
- Allow cleaning procedures to be conducted as required
- Allow sterilization, including sterilization in place (SIP), to be conducted as required
- Permit in-place integrity testing of the sterilizing filter, preferably as a closed system

Qualification/Validation Of Sterile Filters
Sterile filters are usually characterized by their pore size. As already mentioned, the diameter of the pores should be 0.2 µm to 0.22 µm. The specified pore size is "nominal", i.e., the specification is based on values calculated by the manufacturer under ideal conditions. This means that there may also be larger pores in the filter material.
More important for the characterization is the retention capacity, which is influenced by all factors of the filtration process. It is generally assumed that the filtration effect is similar to a sieve function. While the particle size is decisive for the separation effect in the case of geometric, solid particles, other effects also occur in the filtration of microorganisms. Microorganisms and small particles are retained within the pores by adsorptive forces, depending on the following factors:
- Differential pressure
- Flow rate
- Particle count
- Surface tension
- Degree of ionization (adsorption of negatively charged microorganisms and bacterial endotoxins)
A very important point here is the differential pressure, which must not lead to deformation of the microorganisms. If the microorganism is deformed, it can permeate, i.e., be squeezed through the filter.
For sterile filtration the following considerations must be taken into account:
- The greater the number of particles (including microorganisms), the greater the number of particles that are not retained. This means that the bioburden level has a strong influence on the effectiveness of the filtration. This makes it clear that the bioburden should be as low as possible. Determination of the type of bacteria found in the monitoring program is important in order to know the size of the bacteria.
- The lower the differential pressure, the greater the probability of microorganisms being separated.
- The longer the dwell time of an organism in a pore, the more secure its retention.
For the qualification and validation of filters, various aspects must be taken into account. Relevant information on this can be found in EU GMP Guide Annex 1 (paragraphs 8.80 and 8.81) and in the European Medicines Agency’s Guideline on sterilization of the medicinal product, active substance, excipient and primary container. The most important aspects are shown in Figure 1 and are briefly explained.
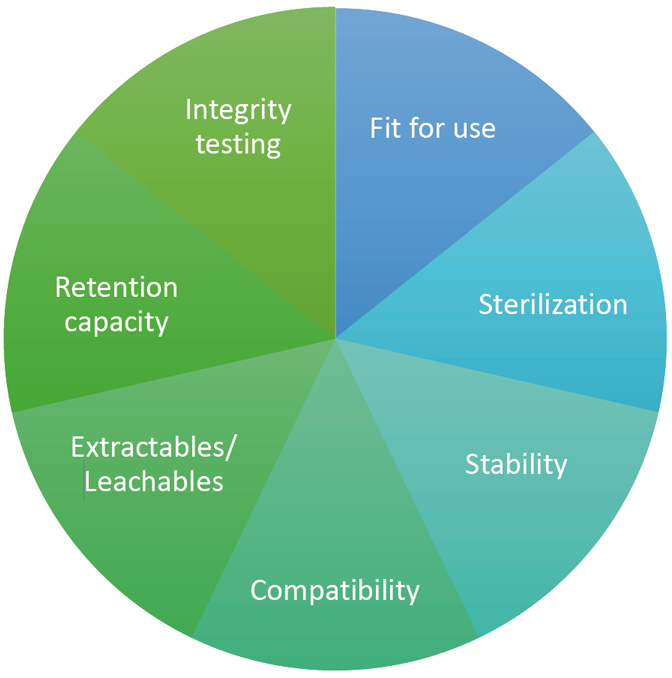
Figure 1: aspects of filter validation
Fit for use
Ensure that the filter meets all requirements within the process and production conditions (e.g., with regard to temperature, pressure, etc.).
Sterilization
Ensure that the sterilization method for the filter is effective and does not damage it.
Stability
Ensure that the filter does not negatively affect the product.
Compatibility
Ensure that the filter is not negatively affected by the product and or adsorbs the active substance.
Extractables/leachables
Identify, quantify, and assess the impact of components that may migrate from the filter into the medicinal product.
Retention capacity
Ensure that the filter can remove microorganisms from the medicinal product.
Integrity test
Ensure that the retention capacity of the filter can be verified with a non-destructive test (e.g., bubble point, pressure retention test, etc.).
Validation Of Sterile Filtration (Bacterial Retention)
Validation of sterile filtration (bacterial retention testing, EU GMP Guide Annex 1, paragraph 8.84) must be carried out in accordance with the relevant pharmacopoeia methods (Annex 1, paragraph 8.83). Worst-case conditions regarding the concentration of the medicinal product in the solution and matrix effects of the solution must be taken into account. The challenge organism used for validation should be well justified. ASTM F838-20 specifies the use of the challenge organism Brevundimonas diminuta for the validation of the retention capacity. This microbe is used due to its small size and its occurrence in water systems. However, it is necessary to check whether the size of existing microbes found on-site would not represent a worst-case scenario. In such cases, the corresponding in-house microbe should be used.
Knowledge of the bioburden, i.e., the microbial load of the solution to be filtered, is important for the following reasons:
- A filter may have some pores that are larger than the nominal pore size, so microorganisms of a certain size may be able to pass through. It is therefore important to know the type of microbes.
- It is assumed that the probability of bacteria passing the filter increases when the number of organisms in the media to be filtered increases, as the load on the filter increases. It is therefore important to know the extent of the bacterial load.
For validation, a concentration of test bacteria of 107 CFU per cm2 filter surface should be present in the product (worst case). Under these circumstances, the filtration should deliver a bacteria-free product. The bioburden is also an IPC monitoring point that must be determined during routine production. For this purpose, a sample of the solution is taken before the sterilizing filter.
Before the actual test, it must be checked whether the product to be tested can be used for validation. A so-called viability test provides information on whether the product has a bactericidal effect. For this purpose, a defined number of test bacteria are incubated in the product solution for the entire duration of the filtration process. A reduction in the number of test bacteria indicates a bactericidal effect of the product. In cases such as these, the use of a surrogate is possible during filter validation (EU GMP Guide Annex 1, paragraph 8.84).
Annex 1 of the EU GMP Guide lists parameters under paragraph 8.85 that should be covered during validation (see Figure 2).
|
EU GMP Guide Annex 1: Manufacture of sterile medicinal products Sterile filtration of products that cannot be terminally sterilized in the final container 8.85 Filtration parameters that should be considered and established during validation include, but are not limited to: i. The wetting fluid used for filter integrity testing:
ii.The filtration process conditions including:
|
Figure 2: EU GMP Guide Annex 1: Validation of sterile filtration
When creating the validation plans it is important to consider the effects of extreme values of all parameters of the production process that may affect the capability of a filter. A risk analysis should be conducted to identify the worst-case parameters and variables. The risk analysis helps to categorize the validation requirements based on the impact on the product, the process, and the individual system components. At the same time, it helps to reduce the overall validation effort.
Using a risk analysis, it is possible to group similar products into product families and carry out validation on just one product. However, this is only possible as long as the same active substance is involved. Different active substances in similar formulations cannot be grouped together.
Sterilization Of Filters
Every sterile filter or system used for sterile filtration must be sterilized before use. This sterilization can take place both inline (i.e., when installed) and offline in an autoclave.
The manufacturer's instructions must be observed during sterilization. Sterilization is normally carried out at 121 degrees C, whereby holding times of 20 to 30 minutes are appropriate. Some filter units can also be sterilized at 134 degrees C or 145 degrees C for 30 minutes.
Steam must flow completely through the filters during sterilization. If this is not fully achieved, air pockets are likely to result. The saturated steam cannot realize its effect in these zones. At these points overheated steam results, which does not have a maximum sterilization effect.
Possible solutions could be:
- Validated preliminary vacuum-cycles are essential for sterilization in autoclaves.
- With SIP, valves must be arranged in such a way that the removal of air pockets by bleeding is safely ensured.
- The inclusion of bioindicators during validation runs is highly recommended.
The effectiveness of the sterilization process of filters in situ is often influenced by condensate formation in the cartridge. The condensate that forms blocks the pores and may prevent the flow of steam through the wetted surface. In this case, the filter heats up unevenly. The solution to this can be slow heating and checking the maintained vapor pressure at the outlet.
Filter Integrity Testing
The integrity (i.e., intactness) of the filter must be checked before and after use. This requirement can be found in Annex 1 of the EU GMP Guide (see Figure 3) and in the European Pharmacopoeia (5.1.1 Methods of preparation of sterile products).
|
EU GMP Guide Annex 1: Manufacture of sterile medicinal products Filtration sterilization of products which cannot be sterilized in their final container 8.87 The integrity of the sterilized filter assembly should be verified by integrity testing before-use (pre-use post-sterilization integrity test or PUPSIT) to check for damage and loss of integrity caused by the filter preparation prior to use. A sterilizing grade filter that is used to sterilize a fluid should be subject to a non-destructive integrity test post-use prior to removal of the filter from its housing. The integrity test process should be validated and test results should correlate to the microbial retention capability of the filter established during validation. Examples of tests that are used include bubble point test, diffusive flow, water intrusion or pressure hold test. It is recognized that PUPSIT may not always be possible after sterilization due to process constraints (e.g. the filtration of very small volumes of solution). In these cases, an alternative approach may be taken providing that a thorough risk assessment has been performed and compliance is achieved by the implementation of appropriate controls to mitigate any risk of a non-integral filtration system. |
Figure 3: EU GMP Guide Annex 1: Filter integrity testing
Particular attention must be paid to the requirement in Annex 1 to perform a filter test on the sterilized filter before it is used. This test, known as PUPSIT, must be included in the process design when developing the filtration process. This requirement is not easy to implement in practice, as carrying out the test must not jeopardize the sterility of the filter and the filter system.
Exceptions to PUPSIT are possible, but must be properly justified and documented with a corresponding risk analysis.
If the sterilizing filtration step consists of multiple filters and validation has been performed for the achievement of sterility of the fluid, the system is considered as a single sterilizing unit and all filters within the system should satisfactorily pass a test for integrity after use (EU GMP Guide Annex 1, paragraph 8.91).
The filter integrity test should be carried out with the filter installed. This can be performed easily with the test devices currently available on the market.
The FDA also requires integrity testing, and it places emphasis on carrying out the integrity test after the filter has been used. Testing for integrity before use remains optional for the FDA. (FDA Guidance for Industry, Sterile Drug Products Produced by Aseptic Processing-Current Good Manufacturing Practice, 2004, Chapter IX Validation of Aseptic Manufacturing and Sterilization, Part B Effectiveness of Filtration.)
As mentioned in Annex 1 of the EU GMP Guide, there are several test methods recommended to check the condition and functionality of membrane filters:
- bubble point test
- pressure decay test
- diffusive flow test, or forward flow test (gas diffusion).
The implementation and special aspects of these test procedures are described in Figure 4.
| Test | Execution/Special Considerations |
|---|---|
| Bubble point test | The bubble point corresponds to the pressure required to push air through the liquid-filled pores of the filter. This pressure can be recognized by a clear stream of air bubbles, hence the name. This pressure depends on the surface tension of the wetting liquid as well as the pore size and the wetting angle of the filter. The test is therefore also dependent on the filter material and the pore size. For 0.2 µm filters, the value is approx. 3.4 to 4.8 bar (cellulose acetate and cellulose nitrate). Knowing the bubble point pressure is important for carrying out the gas diffusion tests. |
| Pressure hold test | During the pressure hold test, pressure is applied to the non-sterile side of the filter (approx. 60% of the bubble point pressure). The pressure must be maintained for 3-5 minutes (within a certain tolerance). If a pressure drop occurs, the filter system is damaged (seals defective, mechanical damage to the filter, etc.). This test is always carried out with the entire filtration system, i.e., filter housing and inlet and outlet. This test is therefore also well-suited for in-process control (PUPSIT). |
| Diffusive flow test/forward flow test | The gas diffusion rate is determined in the forward flow test. To do this, the filter is subjected to 80% of the bubble point pressure. The speed at which the air diffuses through the pores is measured. |
Figure 4: Filter integrity tests
All these tests are highly dependent on the surface tension of the wetting liquid. Different values are therefore obtained when wetting with liquids with different surface tensions, such as water, alcohol, disinfectants, or similar. It should therefore be decided beforehand whether a product-specific integrity test or an integrity test with a standard medium (e.g., water) should be carried out. In the case of a product-specific integrity test, the filter manufacturers are usually the direct point of contact. They can develop a product-specific bubble point test.
Loss Of Filter Integrity
If the filter does not pass the test, there may be several reasons for this:
- Pressure surges (during sterilization or filtration)
- Unsuitable sterilization conditions (e.g., condensation inside the filter cartridge or excessive differential pressure)
- Mechanical damage (e.g., during assembly)
The reason for a filter to fail an integrity test does not always have to be a damaged filter. Alternative causes of a failed test can also be:
- Filters not properly rinsed
- Filters not completely wetted
- Defects in the housing
In order to rule out such errors before the filter is classified as defective, the PDA has established an Integrity Test Troubleshooting Guide in its Technical Report No. 26 (see Figure 5). This provides helpful instructions for ruling out the above-mentioned possible faults. The filter manufacturers often provide their own troubleshooting guides in their instruction manuals. These are adapted to the characteristics of the specific filter.
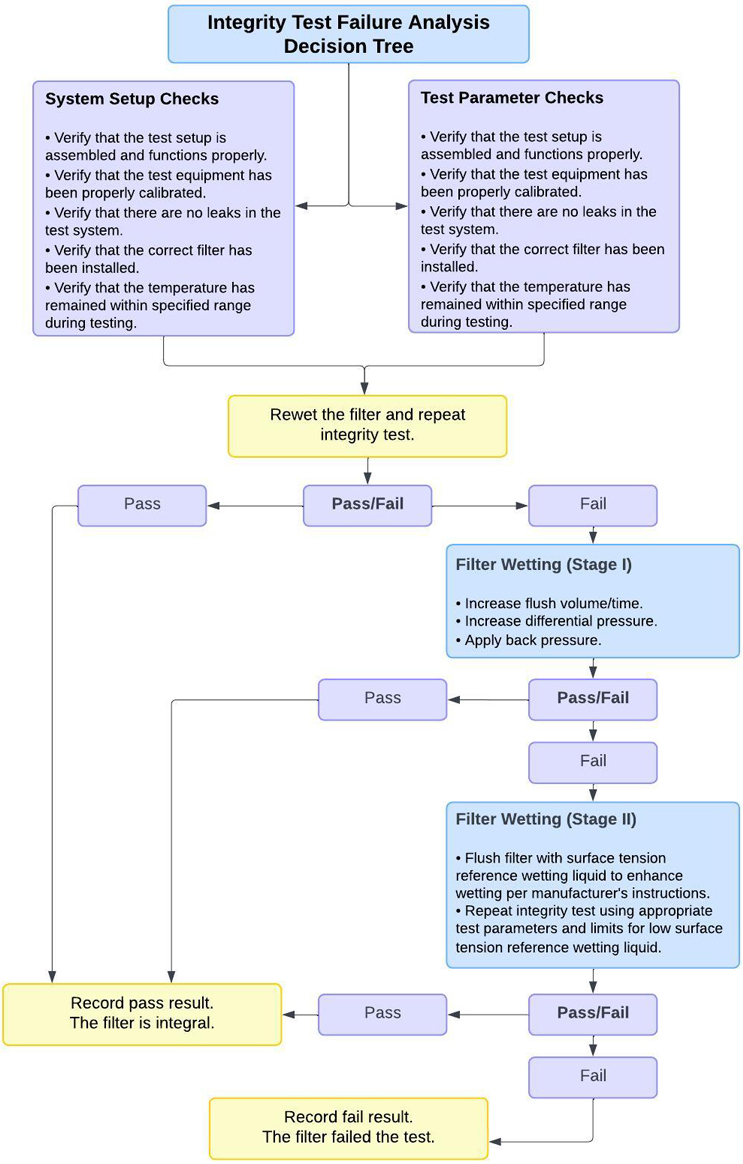
Figure 5: Integrity Decision Tree, Source: PDA Technical Report No. 26
If a filter integrity test fails after filtration, the sterility of the manufactured batch is called into question. As a consequence, the batch can only be destroyed. This applies even if the product sterility test is OK. The sterility test is only a snapshot and is not statistically meaningful.
If a redundant sterile filter system (two sterile filters in a row) is used, the redundant (i.e., the first) filter can be tested. If this redundant filter passes the filter test, the batch can be released in conjunction with a risk assessment and a root cause analysis. However, it is important that only one filter is used for the validation (EU GMP Guide Annex 1, paragraph 8.92).
Reuse Of Filters
Annex 1 of the EU GMP Guide is very clear with regard to the reuse of filters used for final sterilization: use should be limited to one batch and a maximum of one working day (see Figure 6).
|
EU GMP Guide, Annex 1: Manufacture of sterile medicinal products Filtration sterilization of products which cannot be sterilized in their final container 8.94 Liquid sterilizing grade filters should be discarded after the processing of a single batch and the same filter should not be used continuously for more than one working day unless such use has been validated. |
Figure 6: EU GMP Guide Annex 1: Reuse of filters
However, it may be possible to reuse the tested sterile filter as a pre-filter for the same product in order to reduce the bacterial load (bioburden reduction), provided this has been validated. This validation must then also cover the cleaning of the filter. Cleaning filters is very time-consuming and requires significant manpower. There is also a risk of pyrogen accumulation, as the endotoxins of those bacteria retained after re-sterilization and killing are not inactivated. On the other hand, costs can be avoided by reusing sterile filters, especially when producing small batches.
This article is an excerpt from GMP knowledge contained in the online portal GMP Compliance Adviser, which provides in-depth information about GMP best practices and regulations with a focus on Europe, but also referring to the U.S., Japan, and many more (PIC/S, ICH, WHO, etc.).
Editor's note: Several corrections have been made to this article. 1. Per Annex 1, suitable pre-filters and sterile filters "may" be used at multiple points before the final sterilizing filter. A previous version incorrectly said they "must" be used. 2. The text references the ASTM standard for determining bacterial retention of membrane filters used for liquid filtration. The correct standard is ASTM F838-20. 3. Per the ASTM standard, the concentration of test bacteria is 107 bacteria per centimeters squared. In a previous version, the number had been formatted incorrectly. 4. An error in Figure 4 has been corrected to state that pressure in the bubble point test depends upon the "wetting liquid surface tension."
About The Author:
 Raimund Brett works as a principal consultant. In this role, he advises customers from the pharmaceutical and life sciences industry. Cleaning validation, aseptic and sterile production as well as the production of solid dosage forms are the focus of his consulting activities. He also conducts audits of raw material suppliers and contract manufacturers as well as training courses on these topics.
Raimund Brett works as a principal consultant. In this role, he advises customers from the pharmaceutical and life sciences industry. Cleaning validation, aseptic and sterile production as well as the production of solid dosage forms are the focus of his consulting activities. He also conducts audits of raw material suppliers and contract manufacturers as well as training courses on these topics.