
Analytical Challenges In Biosimilar Development
By Mario DiPaola, Ph.D., Scientific Director, and Ulrike Herbrand, Ph.D., Scientific Supervisor, Biopharmaceutical R&D, Charles River
The development of a biosimilar requires the establishment of the quality target product profile (QTPP) early in the development cycle. To develop the QTPP, extensive analytical testing is required, including physiochemical and biological assays, to acquire a thorough understanding of the reference product. The challenge in performing such a task lies in determining the appropriate number of lots to evaluate and ensuring that the chosen lots are representative of the potential product variability and product degradation during storage. Another challenge is the establishment of release specifications, which need to be consistent with the reference product specifications. This paper will describe strategies on how to address these challenges.
Finding Similarity Between Lots
The development process of biosimilars is a stepwise approach in which the biosimilar developer initially focuses on the characterization of the reference product (originator’s drug) with the aim of understanding the product’s quality attributes, including physical, chemical, and biological aspects. While this analytical effort is ongoing, the developer will establish an expression system and begin to produce lots of the biosimilar product for comparison against the reference product. In most cases, the biosimilar drug in development will require some clinical evaluation to assess the pharmacokinetics and pharmacodynamics of the biosimilar product, along with efficacy, as necessary, as well as safety and immunogenicity. If the acquired analytical, animal, and clinical data are found to be similar to the reference drug, the biosimilar developer may ask for regulatory approval of the drug (Figure 1).
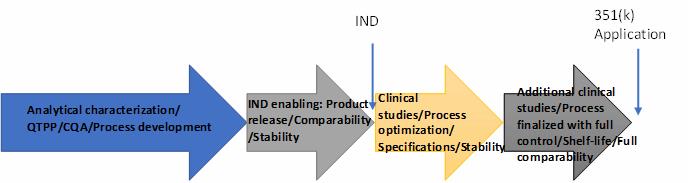
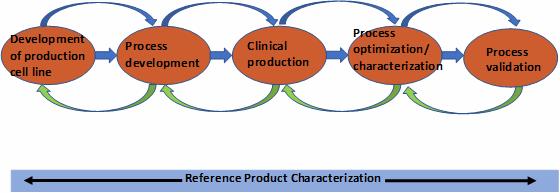
Throughout the development cycle, multiple lots of the reference product are purchased and characterized to obtain as clear a picture as possible of the product’s biological activity, mode of action, primary, secondary, and tertiary (and quaternary, if needed) characteristics, and post-translational modifications (PTMs). Furthermore, the developer will determine how the product PTMs, including oxidation, deamidation, and other stress-induced modifications, alter the biological function(s)/activity and potentially the mode of action. New knowledge gained from the continued reference product testing is then fed into the manufacturing process development to optimize the manufacturing of the biosimilar and introduce sufficient controls to ensure consistent product quality (Figure 2).
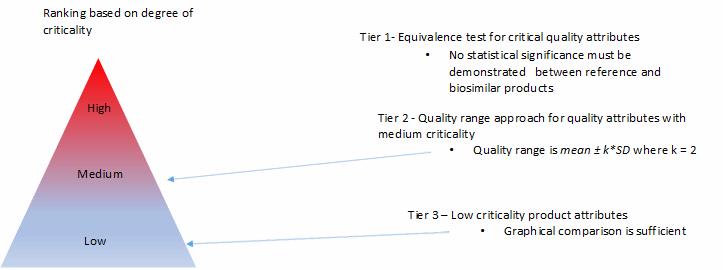
Based on the reference product testing and the established relationship between product attributes and product efficacy and safety, the product attributes are categorized into critical product attributes (Tier 1) that can impact the product safety and efficacy, followed by the less critical attributes (Tier 2), and then noncritical attributes (Tier 3). The rigors of comparability will range from statistical equivalence for Tier 1 attributes to graphical overlay for Tier 3 attributes (Figure 3).
Product specifications are required to be established early in the development cycle to release manufacturing lots for use in analytical comparability and animal/clinical studies and to assess biosimilar stability. The early product specifications to enable product release and stability will be mostly based on analytical data accumulated from the analysis of the reference product and
with some considerations of process capabilities at the time of specification establishment. It should be noted that when selecting reference product lots for analytical characterization, the chosen lots should span the entire product stability duration (shelf-life). This is necessary to ensure that the specification limits capture not only manufacturing process variability but also variability due to product instability during storage.
Furthermore, if the biosimilar is aimed for global distribution, the product lots should be picked out from different geographies. It is recommended that data from a minimum of 10 lots of the reference product be used to set the initial product specifications; of course, as more data becomes available from the analysis of additional reference product lots, the established specifications should be revisited.
While the analytical data from the reference product is going to drive the specification-setting process for the biosimilar in the early and middle stages of the development cycle, for 351(k) filing and in support of commercial product release, the biosimilar product specifications should reflect the finalized biosimilar manufacturing process capabilities.
Multiple MoAs For One Antibody
Another challenge in early-stage biosimilar development is defining the target product profile, which drives the identification of the critical quality attributes (CQAs).
According to ICH Q8, a CQA is a physical, chemical, biological, or microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired product quality.
The in vivo mechanism of action (MoA) — the specific biochemical interaction that enables a drug to work — is one of the key drivers for the identification of the CQAs. For complex protein therapeutics like monoclonal antibodies, which are currently a major focus in biosimilar development due to patent expiry of the first anti-cancer and anti-inflammatory commercial monoclonal antibody (mAb) therapeutics, identifying the MoA becomes complicated because more than one MoA is often relevant for efficacy. This means all potential MoAs need to be addressed during in vitro analytical characterization in order to meet regulatory requirements and ensure future patient safety.
Many of the first-generation anti-cancer mAbs follow the so-called classical immunological pathways via Fc gamma receptors, with MoAs including antibody-dependent cellular cytotoxicity (ADCC) by activation of the patient’s natural killer (NK) cells and antibody-dependent cellular phagocytosis (ADCP) by activation of the patient’s macrophages, or by activating the patient’s complement cascade via C1q binding and inducing complement dependent cytotoxicity (CDC). Other full-length mAbs, like the immune checkpoint mAbs, are capable of activating the patient’s T-cell response as the major MoA.
As long as full-length mAbs require characterization, including those that bind soluble targets, the classical immunological pathways must be included in the characterization strategy as potential secondary MoAs, as these might play a role in the overall efficacy of the product and the similarity between innovator and the proposed biosimilar that must be demonstrated.
For thorough characterization of the biosimilar, a reasonable number of approved innovator lots representing different markets and production sites must be evaluated. And the number of lots chosen for analysis should be driven by availability and the variability of the applied methods.
A major goal in the initial characterization of the reference product should be the linking of physicochemical result to bioassay results. Gaining an understanding of the links between chemical properties and bioactivity is important for the determination of critical quality attributes which, once established, will guide the manufacturing process development and optimization.
Surrogate Vs. Traditional Assays
A further major challenge in analyzing antibodies is finding their sweet spot. In mAb therapeutics, there are several regions within the molecular structure of these molecules involved in defining the biological activity.
The antibody is specific for its target, be it a cell surface receptor or soluble ligand, through the antigen binding region in the Fab domain, which recognizes a unique epitope on the target. So, location matters. The correct choice of methods to determine the epitope binding site associated functions as well as potential and known Fc-mediated effector functions is of great importance to the success of a biosimilarity program and should be based on several aspects. The methods should closely reflect the in vivo situation, suggesting that primary cell-based assays, including cell types relevant for the in vivo response, should be considered. However, such methods tend to be tedious and highly variable, leading to significant challenges with data interpretation.
Furthermore, it should be noted that even the use of different cell lines or different readouts addressing the same MoA might lead to different results, implying that the choice of method needs to be carefully evaluated. Different results are not necessarily indicative of a faulty method but may simply be indicative of varying capabilities and limitations of individual methods, cell lines, or readouts to detect differences between products. The acceptance of test methods based on engineered cell lines, including reporter constructs (so-called surrogate assays), is increasing significantly and might be the preferred option to move forward. Additionally, regulatory bodies are becoming more accepting of surrogate biological methods as they recognize the limitations of traditional methods.
Choosing the bioassay(s) for demonstration of biosimilarity should be a case-by-case decision, and aspects like sensitivity and specificity should always be taken into consideration. In some cases, the pathway of choice is to include primary cell-based data for characterization purpose, as supportive data, and then switch to the surrogate methods for biosimilarity assessment between innovator and biosimilar lots. This strategy will allow less work up front, since assays for characterization require only qualification, whereas methods that are intended for later-stage lot release and stability testing require a full validation.