
CDMOs And Contamination Control Strategy: The Span Of Oversight In EU GMP Annex 1 Compliance
By Ryan Murray, M.Sc., and Amanda McFarland, M.Sc., ValSource, Inc.

With the August 2022 released revisions to Annex 1 of the EMA's Eudralex Volume 1 and the increased expectations it brings to sterile drug producers, CDMOs are finding their sterility assurance programs in a position that will require a new level of transparency with sponsors. Under the new set of requirements, the "span of oversight" has shifted and in most cases, this shift benefits the CDMO's sponsor. CDMOs have often kept information related to their sterility assurance programs somewhat reserved, to ensure confidentiality of client-specific information. However, the revision to Annex 1 now requires a documented, robust assessment of the process, facility, and equipment related to sterile manufacturing to ensure that a contamination control strategy (CCS) can be implemented, followed, and monitored. Alongside the needed transparency of the CCS, additional challenges for CDMOs that arise in the face of Annex 1 include potential facility design and equipment changes, workforce shortages, expertise to champion the CCS effort, quality agreement modifications, and differing CDMO-sponsor interpretation of topics included in the Annex, such as pre-use post-sterilization integrity testing (PUPSIT) of drug product filters.
Contamination Control Strategy
One of the most broad and impactful elements of the Annex 1 revision is the concept of a contamination control strategy. While the concept of contamination control is not new, the requirement for a formally documented strategy specific to a facility is a new and key requirement of the revision. The contamination control strategy is intended to be a singular document that serves as a point of reference for the product and process critical control points and a "one stop shop" for demonstrating how a state of control is scientifically established, maintained, and remains effective. Strongly supported by quality risk management (QRM) principles, the CCS will draw upon the bodies of knowledge contained within the risk assessments that help inform the process controls to protect product quality and patient safety. The client–CDMO relationship in the context of information sharing can feel a bit like an opportunity to learn that secret cookie recipe you've been wanting to know. Sponsors provide CDMOs a private baking lesson by granting access to their know-how, process, and technology trade secrets, training insights, and other confidential information — they are then relying on the CDMO to execute the "recipe" in a controlled environment and define, with specificity, the state of control such that the cookie is indistinguishable from the original kitchen's. In exchange for the "secret recipe" from an existing client, the CDMO must also share elements of the CCS that are indirectly related to the client's product and process. CDMOs also have the challenge of providing this visibility to a prospective client without compromising an existing client's confidential information and process intellectual property. Here, it may be beneficial to diverge from the proposed table of contents outlined in Annex 1 and utilize a strategy that separates the process and client-specific controls into dedicated appendices.
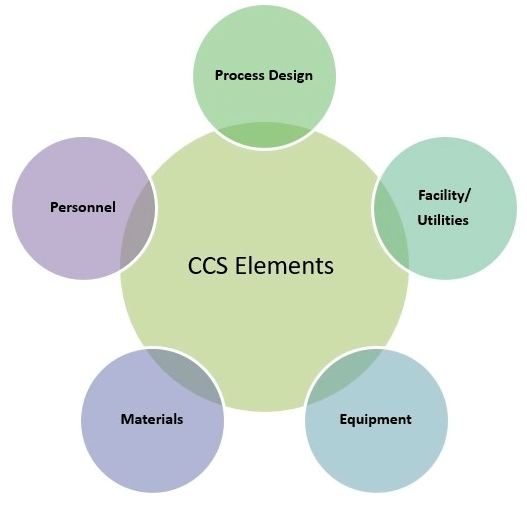
Determining the boundaries of the sponsor–CDMO relationship as it pertains to CCS is critical. Consider Figure 1: Each of these elements is commonly assessed through QRM activities and managed to demonstrate a state of control.
Figure 1. Contamination Strategy Control Elements
To successfully fulfill the CCS requirement as a CDMO, it will become necessary to develop a plan that outlines the span of oversight illustrated in Figure 2. The assessments pertaining to personnel, utilities, and facility can be approached from a product/process agnostic perspective. Inputs related to the materials and equipment will be a collaborative exchange between the CDMO and sponsor, while the controls relative to the process, such as the critical process parameters in place to control quality attributes, will be largely provided by the sponsor. When considering the span of control from each party, the bolus of the CCS documentation will live with the CDMO, with careful consideration of the way that data is collated such that information can be shared seamlessly with the sponsors.
Figure 2. CCS Span of Oversight
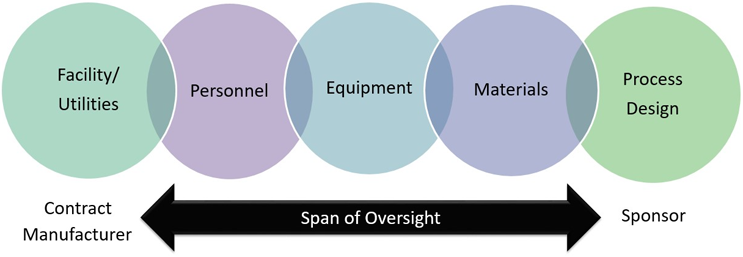
In addition to sharing the product/process agnostic CCS elements with a client as demonstrated in Figure 2, an element of this reciprocity for "secret recipe" sharing will likely be changes to established quality agreements. From the perspective of a client, a CDMO may play only one part of the production operation; they may produce the drug substance, drug product, an intermediate, be a critical material supplier, or any combination thereof. As the client of a CDMO, these pieces are woven together to define product sterility risks from end-to-end. Whether the CDMO plays the role of supplier of a critical intermediate, drug substance producer, or drug substance or fill/finish manufacturer, the control points for enabling sterility assurance at the CDMO must be fully understood by the client. Therefore, additional contractual requirements for communicating points of failure of the sterility assurance program should be clarified the quality agreement. A key area where this exchange of information occurs is during joint risk assessments.3 The time and resources needed to execute these assessments should be documented in the quality agreements.
Talent Acquisition And Retention
To ensure they can meet the rigorous requirements of Annex 1, CDMOs will need to evaluate the staffing and resources required to develop and maintain the CCS. A point addressed in our Annex 1 Roadmap (September 2022). CDMOs have been heavily impacted by the labor shortages of recent times, and the increased expectations of sterility assurance controls established by the revision to Annex 1 will add to the workload of a CDMO. Therefore, CDMOs will need to be particularly strategic in their path to compliance. And it is not just a lack of people that presents the issue. The expertise needed to fill the roles of sterility assurance stewards and aseptic operators includes extensive onboarding programs as well as documented controls for the qualification and disqualification of personnel. In the realm of staffing shortages within CDMOs, it is a frightening reality that the disqualification of an operator could lead to a drug delivery interruption. Furthermore, an expanding and contracting workforce runs the risk of negatively impacting the organization's quality culture. A mass exodus of knowledge and rapid onboarding can often result in a shift in culture. Management and human resources should work together to ensure that a consistent message is sent to all employees that reinforces the quality standard. Leadership should define a proactive plan to monitor and address shifts in behaviors, such as through transparency, collaboration, and accountability.
Conclusion
To deliver safe and effective medicines to our patients, Annex 1 is driving us toward a new standard. It is a paradigm in which sterile drug sponsors and their contract manufacturers can no longer exist in isolation and practices for assessment, control, and monitoring for sterility assurance must be shared. The implementation deadline for the Annex 1 revision is August 2023 and now is the time to determine if the existing CDMO–sponsor relationship is sufficient to enable successful adherence to Annex 1. Establishing how each organization contributes to the safety of patients will facilitate conversations that are focused on the big picture. As staffing shortages linger, the margin for failure shrinks. CDMOs should take responsibility for the trust extended by the sponsor in sharing their recipe and take time to sketch out the CCS and identify expectations related to risk assessment execution and transparency. The mutual acknowledgement of the span of oversight will enable successful production of sterile products and facilitate stronger relationships between sponsors and CDMOs.
About The Authors:
 Ryan Murray is a quality and manufacturing science senior consultant with ValSource, Inc. His focus is in the areas of facility design and control, technology transfer, process qualification, and aseptic risk management in biologics and advanced therapy medicinal products (ATMPs). He is an active member of the International Society for Pharmaceutical Engineering (ISPE) and the Parenteral Drug Association (PDA) and holds a B.S. in biomedical science and an M.S. in biochemistry and biophysics from Texas A&M University. He can be reached at rmurray@valsource.com.
Ryan Murray is a quality and manufacturing science senior consultant with ValSource, Inc. His focus is in the areas of facility design and control, technology transfer, process qualification, and aseptic risk management in biologics and advanced therapy medicinal products (ATMPs). He is an active member of the International Society for Pharmaceutical Engineering (ISPE) and the Parenteral Drug Association (PDA) and holds a B.S. in biomedical science and an M.S. in biochemistry and biophysics from Texas A&M University. He can be reached at rmurray@valsource.com.
 Amanda McFarland is a quality risk management and microbiology senior consultant with ValSource, Inc. She specializes in the creation and implementation of risk management programs and developing risk-based strategies for use in clinical and commercial settings. McFarland is an active member of the Parenteral Drug Association (PDA), co-chair of the PDA ANSI standard on QRM in aseptic processing, a member of the PDA Regulatory Affairs and Quality Advisory Board, and an instructor for the PDA. She has a B.S. in entomology and an M.S. in mycology, both from the University of Florida. She can be contacted at amcfarland@valsource.com.
Amanda McFarland is a quality risk management and microbiology senior consultant with ValSource, Inc. She specializes in the creation and implementation of risk management programs and developing risk-based strategies for use in clinical and commercial settings. McFarland is an active member of the Parenteral Drug Association (PDA), co-chair of the PDA ANSI standard on QRM in aseptic processing, a member of the PDA Regulatory Affairs and Quality Advisory Board, and an instructor for the PDA. She has a B.S. in entomology and an M.S. in mycology, both from the University of Florida. She can be contacted at amcfarland@valsource.com.