
Considerations For Robust Implementation Of The Multi-Attribute Method
By Diane McCarthy and Li Jing, United States Pharmacopeia

Any analytical method is only as good as the consistency and reliability of its output.
A successful multi-attribute method (MAM) relies on the performance of different components such as sample preparation, instrumentation, and software and careful method design and implementation to ensure robust, consistent results.
In part 1 of this series, we discussed applications and advantages of MAM in process development and quality control. Here, in part 2, we’ll explore implementation strategies, including best practices summarized in USP General Chapter <1060> Mass Spectrometry-Based Multi-Attribute Method for Therapeutic Proteins.
Sample Preparation
Consistent sample preparation is critical to design and develop a reliable MAM with minimal inter-assay variation and sample preparation-induced modifications and artifacts.1 Because MAM evaluates the profile of molecular variants within a preparation, it is critical to reduce artificial modifications that can be introduced during sample preparation. Undesirable modification of the analyte can be introduced during the digestion process and interfere with detection and quantitation of protein modifications.
A typical MAM workflow uses a reduced peptide mapping workflow for the relative quantitation of post-translational modifications (PTMs) such as oxidation and deamidation. A typical digestion workflow (Figure 1) includes denaturation, reduction, alkylation, and enzymatic digestion. The pH and temperature can impact modifications and need to be assessed during method development. Sufficient incubation time is required to achieve complete denaturation, reduction, alkylation, and, subsequently, digestion, but longer processing times can lead to an increase in method-induced artifacts. Automation is often used to reduce variability and improve efficiency in sample preparation.2 Options and technical considerations for each step of the sample preparation procedure are provided in USP General Chapter <1060>.
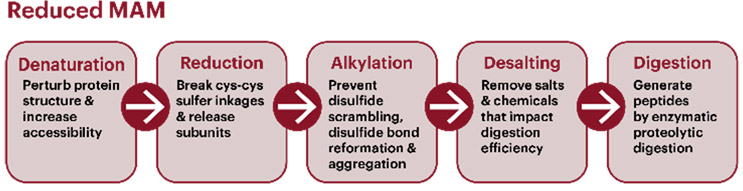
Figure 1. Schematic of a sample preparation workflow for a reduced MAM. The five-step process starts with breakdown of quarternary, tertiary, and secondary structure elements of the protein of interest. For mAbs, it is critical to break all disulfide linkages (Cys-Cys) with a reducing agent and to prevent reformation with alkylating treatment. Samples should then be desalted to remove any impurities that can impact the efficiency of enzymatic digestion.
System Suitability And Readiness
A successful method requires a fit-for-purpose MAM system whose readiness can be verified. System readiness indicates that the analytical system is functioning and passes predefined criteria, making it ready for analysis. In cGMP testing, a formally defined system suitability test should be established. A robust system readiness MAM method can simultaneously evaluate the performance of sample preparation, instrumentation, and software. Checks on the individual components of the MAM system may be desired or required prior to system readiness check.
System readiness metrics should cover a range of system performance parameters and be sensitive enough to alert users to possible problems. Common system readiness metrics are listed in Table 1. Well-characterized standards are needed to support the system readiness assay and can include peptide mixtures, commercial proteins, and in-house materials. Advantages and disadvantages of different standards, as well as more detailed guidance on establishing system readiness, are also provided in USP General Chapter <1060>. USP also offers a suite of well-characterized reference standards to support MAM analysis of mAbs as discussed below.
| Attribute | Criteria |
|---|---|
| Total ion chromatography (TIC) signal intensity | Above specific threshold |
| Mass accuracy | Within specified range |
| MS resolution | Within specified range at a given m/z |
| Retention time | Within specified range |
| Chromatographic resolution | Within specified range measuring between two peaks |
| Integrated peptide area | Within specified range |
| Methionine oxidation | Within specified range |
| In-source fragmentation | Below specified range |
| MS/MS fragment ion intensity | Above specified range |
| MS/MS fragment ion mass accuracy | Within specified range |
Table 1. Common system readiness metrics.
Application Of MAM For Process Development
MAM may be one of the most revolutionary tools for strengthening the understanding of the relationship among manufacturing process steps, protein modifications, and function. Site-specific multiple PQA data can provide a more detailed view of protein modifications that can occur during expression and purification of a protein compared to data generated by conventional methods.
Among the applications of MAM for process development is the assessment of deamidation sites for criticality. Figure 2 compares the degradation profiles measured by MAM and icIEF of a mAb produced by two different processes and maintained at 40°C for 18 weeks. Changes in the levels of charge variants were monitored by each method. Because icIEF and other charge variant assays typically do not have the resolution to electrophoretically separate every charge variant, changes in the level of acidic charge variants are reported as a single non-specific value (% acidic). In contrast, MAM enables direct identification and specific monitoring of individual product quality attributes affecting the charge variant profile and allows identification of where those changes occur within the protein.

Figure 2. Thermal stability of a mAb manufactured using two different processes. This shows the relative abundance of all acidic charge variants as determined by icIEF (left) and six specific deamidation sites (1-6) monitored by MAM (right). Adapted from General Chapter <1060> Mass Spectrometry Based Multi-Attribute Method for Therapeutic Proteins
The MAM data reveal how process changes affected the levels of deamidation at six specific asparagine or glutamine sites (Figure 2). Two of the residues with increasing deamidation were in the complementarity-determining region (CDR) of the light chain, which is important for proper binding of the antibody to its target; the other deamidation site was a non-CDR region of the light chain. Three significant deamidations occurred in the Fc-region of the heavy chain and may affect Fc-mediated effector functions of the mAb, such as antibody-dependent cellular cytotoxicity (ADCC). Rather than setting specifications and controls for the total percentage of acidic species, MAM enables each deamidation site to be assessed for its own criticality in terms of function and safety. MAM can be used for a variety of other applications during process development including assessing modifications induced by thermal stress, assessing modifications that are impacted by formulation, and assessing the effect of bioreactor and process conditions on the protein.

Implementation Of MAM In The QC Environment
In addition to its use for characterizing PQAs during process development, there is a growing interest in using MAM as a release test to possibly replace several other methods. MAM is well-suited for this application because it can simultaneously measure multiple quantitative CQAs. It can also detect new peaks that may appear during the manufacturing, storage, or handling of the drug product. There are four key considerations for implementing MAM as a QC method in the cGMP environment.3 These include conducting a risk assessment of implementation, comparing its performance with conventional methods, validating the method, and understanding the capabilities and specificities of the new peak detection (NPD) feature. However, broader adoption of MAM in the QC environment will also rely on standardization and the development of best practices to ensure consistency across multiple sites and organizations.
USP Standards And Tools To Support Characterization Of Biotherapeutics Using MAM
Given the opportunities and benefits offered by MAM, USP has developed standards and scientific content to support its adoption. The proposed USP General Chapter <1060> was published in September 2023 for public comment in Pharmacopeial Forum. This chapter was developed by a USP Expert Panel of MAM experts from industry and is designed to help drug developers and manufacturers explore this exciting new approach to analysis of proteins. The chapter provides detailed guidance for its implementation both in the process development and QC setting and for transitioning from conventional assays (Figure 3) to MAM. The chapter includes extensive information on sample preparation, system readiness, validation, and NPD.
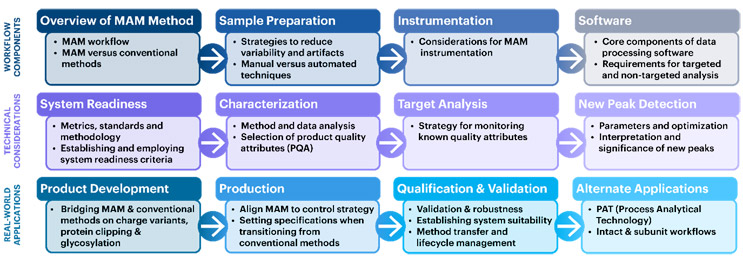
Figure 3. USP General Chapter <1060> details sample preparation, system readiness, general considerations, and case studies for MAM. Guidance on assay validation, method transfer, and implementation in QC are also provided.
Along with publishing the new chapter on MAM, USP also offers several monoclonal antibody standards that can be used to establish system suitability for various analytical methods including MAM. Pre-digested versions of these mAbs are in development and will enable users to assess both instrumentation and the method itself for peptide mapping and MAM applications. Through a cooperative agreement with FDA, USP is also conducting studies to compare conventional methods to MAM using adalimumab and etanercept as examples of mAb and fusion protein therapeutics.* This data set will be made publicly available when the study is completed and can help facilitate adoption of MAM.
MAM is an exciting new method for quantitatively testing and monitoring PQAs during process development and can offer significant advantages when implemented as a QC method as part of the control strategy for therapeutic proteins. The solutions provided by USP can facilitate more rapid and efficient adoption and implementation of this powerful analytical method to derive more detailed insight into bioprocesses and products.
* Acknowledgement of Federal Support: This project is supported by the Food and Drug Administration (FDA) of the U.S. Department of Health and Human Services (HHS) as part of a financial assistance award FAIN# U01FD007762 totaling $1,530,721 with 100% funded by FDA/HHS. The contents are those of the author(s) and do not necessarily represent the official views of, nor an endorsement, by FDA/HHS or the U.S. Government.
References:
- Guo J., Mugabe S., Tu H., Jing L., Peckham N., McCarthy D., Atouf F. (2022) Impact of Sample Preparation Conditions on Detection of Post-Translational Modifications using a Multi-Attribute Method Workflow. Poster presented at CASSS meeting.
- Millán-Martín S., Jakes C., Carillo S., Buchanan T., Guender M., Kristensen D.B., Sloth T.M., Ørgaard M., Cook K., Bones J. (2020) Inter-laboratory study of an optimised peptide mapping workflow using automated trypsin digestion for monitoring monoclonal antibody product quality attributes. Anal. Bioanal. Chem. 412(25), 6833–6848
- Rogstad S., Yan H., Wang X., Powers D., Brorson K., Damdinsuren B., Lee S. (2019) Multi-Attribute Method for Quality Control of Therapeutic Proteins Analytical Chemistry 91(22), 14170-14177
About The Authors:
 Diane McCarthy, Ph.D., is senior director, science and standards in USP’s Global Biologics Department, where she leads development and maintenance of standards and tools to support quality of medicines and oversees the USP biologics laboratories in the U.S. and India. Her team supports standards and tools across a diverse range of therapies, including vaccines, peptides, cell and gene therapy, monoclonal antibodies, and other protein therapeutics. Prior to joining USP, Diane worked for several small CROs that focused on the use of mass spectrometry for characterization of biologics, host cell proteins, and biomarkers. McCarthy earned her Ph.D. in biochemistry from the University of Texas at Austin.
Diane McCarthy, Ph.D., is senior director, science and standards in USP’s Global Biologics Department, where she leads development and maintenance of standards and tools to support quality of medicines and oversees the USP biologics laboratories in the U.S. and India. Her team supports standards and tools across a diverse range of therapies, including vaccines, peptides, cell and gene therapy, monoclonal antibodies, and other protein therapeutics. Prior to joining USP, Diane worked for several small CROs that focused on the use of mass spectrometry for characterization of biologics, host cell proteins, and biomarkers. McCarthy earned her Ph.D. in biochemistry from the University of Texas at Austin.
 Li Jing, Ph.D., is a senior manager in USP’s Global Biologics Department, where she leads a team working with the USP expert committees and multiple expert panels for proteins, peptides, and carbohydrates to develop standards that support biopharmaceutical quality assessment and development. Recently, Jing worked with the USP MAM Expert Panel and developed General Chapter <1060> Mass Spectrometry Based Multi-Attribute Method for Therapeutic Proteins. Jing holds a Ph.D. in analytical chemistry from the University of Georgia and a B.S. in chemistry from Fudan University. She has worked for several biotechnology and pharmaceutical companies, focusing on the development of protein therapeutics and vaccine candidates.
Li Jing, Ph.D., is a senior manager in USP’s Global Biologics Department, where she leads a team working with the USP expert committees and multiple expert panels for proteins, peptides, and carbohydrates to develop standards that support biopharmaceutical quality assessment and development. Recently, Jing worked with the USP MAM Expert Panel and developed General Chapter <1060> Mass Spectrometry Based Multi-Attribute Method for Therapeutic Proteins. Jing holds a Ph.D. in analytical chemistry from the University of Georgia and a B.S. in chemistry from Fudan University. She has worked for several biotechnology and pharmaceutical companies, focusing on the development of protein therapeutics and vaccine candidates.