
Could Naïve Patients Be The Key To U.S. Biosimilar Success?
By Terri Stewart, Abraxeolus Consulting

Early in my career, I spent a great deal of time thinking about what the biosimilar market of the future would look like. Of course, I referred to these medicines as “biogenerics” at the time and earnestly believed the new pathway would result in deep savings and increased patient access. Perhaps I was naïve to believe the very issues that had plagued the utilization of and patient confidence in the traditional generic medicines market were history, lessons learned. A decade after the primary negotiations that resulted in the Biologics Price Competition and Innovation Act (BPCIA), it is safe to say the promise of biosimilars is yet to be realized. And as it turns out, perhaps naivety, in a clinical sense, is the solution.
In Europe, over the past 10 years, biosimilars have had a very different trajectory. Since the first biosimilar, somatropin, was approved in 2006, there have been more than 700 million days of patient experience with EU-approved biosimilar medicines.1 There is clearly a level of acceptance by patients and prescribers leading to continued utilization as biosimilars enter the market. Many attribute this to a number of factors, including confidence in health authorities to determine efficacy. In many ways it is difficult to contrast the U.S. and EU pharmaceutical markets given their intricacies, however, the ability of the government to direct utilization of lower-cost alternatives is key to initial patient use across these systems.
So, how then does the U.S. get patients to trust and, more importantly, use biosimilars? One of the difficulties, of course, is inherent in the BPCIA, a statute that sets a standard for biosimilar and interchangeable biosimilar medicines. Some would have you believe this establishes a two-tiered system, with biosimilars being a lesser class of medicines than interchangeable biosimilars. We know interchangeability can be requested upon submission of the initial biosimilar application, and sponsors have sought the designation. To date, the FDA has yet to approve an interchangeable biosimiliar therapy. While the timing of final guidance and first interchangeable approval are unknown, this has not stopped the FDA from accelerating its efforts to capture more patient days.
The administration has carefully choreographed a series of actions to increase the utilization of biosimilars, and it appears its focus is largely directed at new patients:
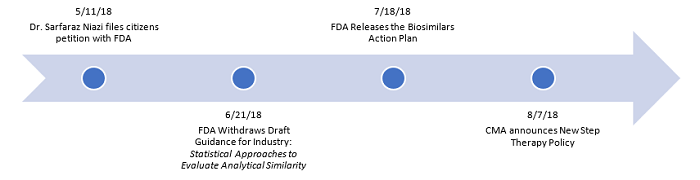
The FDA has acknowledged it is taking into consideration the comments made by Dr. Sarfaraz Niazi in his Citizen Petition to the agency, which suggested modifications to the agency’s policies to expedite the use of biosimilars. Within the petition, Niazi makes a passionate case for a focus on naïve patients. He believes “there is an unmet need to create a new image and status for biosimilars licensed by the agency,”2 acknowledging the difficulty the duality of biosimilar approvals presents. His recommendation is clear that “from a scientific and clinical view point, if a product is clinically equivalent, there is no reason it cannot be used for use in naïve patients.”3
It’s an elegant and science-based solution to one of the fundamental challenges biosimilars face. The only way to fundamentally overcome negative perceptions about biosimilar therapies it to have documented patient success. Instead of winning over patients and prescribers who have already initiated a therapy or delaying greater access while switching studies are conducted and final guidances drafted, focus on building confidence with new patients. This isn’t going to be accomplished through direct-to-consumer marketing of biosimilars, but rather by utilizing the tools that have yielded success in the past.
When we look at the challenges faced by traditional generic drugs, we find some corollaries. There were long-standing policies that undermined confidence in the FDA approval process and the “sameness” of generics to the brand drugs they referenced for the basis of their approval. Much like with biosimilars, states passed legislation that limited the use of generics and placed them on narrow therapeutic index (NTI) lists, making substitution more difficult. However, as prescription drug prices rose and Medicare Part D plans found success in managing costs by maximizing generic substitution, use of generics increased and public perception about generics changed. Today, generics account for nearly 90 percent of all prescriptions in the U.S.4 It is difficult for most of us to imagine what the cost of medicines would be had the perception of generic medicines not changed.
Just last month, when CMS announced its policy to allow the use of step therapy in Medicare Advantage plans, it took important action to change the perception of biosimilars. Under this policy, patients can be initiated on a biosimilar and then moved to a reference biologic only if the treatment has failed. This policy is often referred to as “fail first” and is a tool that has been used previously in formulary management to contain costs. But perhaps the most significant aspect of this policy change is that it will begin to increase the patient treatment days for biosimilars. It is nearly impossible to refute positive patient outcomes, and when coupling positive results with even moderate savings, the trend lines will only point to greater biosimilar utilization. Given these dynamics and the focus of the administration on biosimilars, there is significant opportunity for biosimilars to finally reach their true potential in the U.S.
In fact, the timeline of recent events gives us a few important takeaways about the current health policy developments in biosimilars: 1) The FDA is committed to the biosimilar market and is open to scientific information from industry experts to shape its actions; 2) The FDA is closely coordinating with CMS and other agencies to maximize the effects of their policy ideas; and 3) The FDA, CMS, and the administration writ-large are so confident the outcomes for patients taking biosimilars will be positive, they have made it a cornerstone of their policy actions. These three developments could very well lead to naïve patients using biosimilars more frequently. As we have seen in Europe, positive patient experiences, along with government programs promoting the use of biosimilars, are vital for success. Yes, a new image is truly needed in the U.S. for biosimilars, and the administration’s determination as well as focus on naïve patients will be the root of its creation.
References:
- Medicines for Europe, Factsheet on Biosimilar Medicines 2016.
- Dr. Sarfaraz Niazi, Citizen Petition to FDA, RE: The Biologics Price Competition and Innovation (Subtitle A) of Title VII—Improving Access to Innovate Medical Therapies (H.R. 3590), where Sec. 7002 details “Approval Pathway for Biosimilar Biological Products.” May 11, 2018.
- Ibid.
- AAM 2017 Generic Drug Access and Savings in the U.S. Report.
About The Author:
 Terri Stewart is senior advisor of Abraxeolus Consulting. She offers problem solving leadership and expertise in healthcare in areas such as government affairs, regulations, policy, and their direct implications for a pharmaceutical firm’s operations. Stewart has had extensive interaction with Congress, federal agencies, trade associations, and healthcare industry leaders with the proven ability to drive consensus. Previous roles include VP of global regulatory intelligence, policy, and compliance for Teva Pharmaceuticals and lead federal lobbyist for Barr Laboratories. She holds a degree in government affairs from George Mason University and a J.D. from Catholic University’s Columbus School of Law. You can reach her at Terri.Stewart@abraxeolus.com or connect with her on LinkedIn.
Terri Stewart is senior advisor of Abraxeolus Consulting. She offers problem solving leadership and expertise in healthcare in areas such as government affairs, regulations, policy, and their direct implications for a pharmaceutical firm’s operations. Stewart has had extensive interaction with Congress, federal agencies, trade associations, and healthcare industry leaders with the proven ability to drive consensus. Previous roles include VP of global regulatory intelligence, policy, and compliance for Teva Pharmaceuticals and lead federal lobbyist for Barr Laboratories. She holds a degree in government affairs from George Mason University and a J.D. from Catholic University’s Columbus School of Law. You can reach her at Terri.Stewart@abraxeolus.com or connect with her on LinkedIn.