
FDA Approval Of The First nr-AxSpA Biologics: Projecting Regulatory & Market Dynamics
By Maria Genco, Ph.D., Decision Resources Group (DRG)

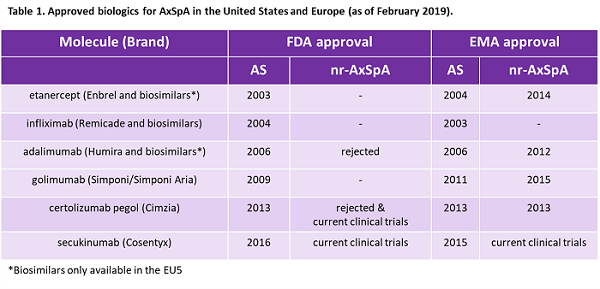
Spondyloarthropathies are a family of inflammatory diseases including psoriatic arthritis and axial spondyloarthritis (AxSpA) that display peripheral or axial arthritis and share common clinical and genetic features, including the genetic risk factor HLA-B27. AxSpA is a chronic disease characterized by axial arthritis that manifests as lower back pain.1-3In advanced cases, it leads to new bone growth resulting in fusion of the spine and significant loss of movement. Physicians have long recognized ankylosing spondylitis (AS), a form of AxSpA where sacroiliitis (inflammation of the sacroiliac joints located at the lower part of the spine) is visible in X-rays. The modified New York (mNY) criteria, developed in 1984, is commonly used for diagnosing AS and, crucially, requires radiographic sacroiliitis. TNF-α inhibitors and one IL-17 inhibitor are currently approved for AS in Europe and the United States (Table 1), and these biologics, used after failure of first-line NSAIDs, are widely regarded as effective for AS.
Over time, physicians have increasingly recognized a non-radiographic form of AxSpA (nr-AxSpA) where inflammation of sacroiliac joints can be detected using MRI but not by X-ray.4 The natural history of the disease is unclear, as some patients with nr-AxSpA go on to develop radiographic sacroiliitis (and a diagnosis of AS) while others spontaneously remit or never develop radiographic changes.2 Interviewed rheumatologists recognize this lack of understanding as a critical unmet need that often leads to delayed diagnosis.5,6Even specialists can find nr-AxSpA difficult to recognize, a challenge compounded by known inter-reader variability when diagnosing sacroiliitis by X-ray or MRI.7 In addition, about 80 percent of American adults will experience non-inflammatory lower back pain sometime during their lives, and rheumatologists seek to avoid misdiagnosis that could lead to unnecessary treatment with biologics.8
In 2009, the Assessment of SpondyloArthritis International Society (ASAS), recognizing an unmet need in nr-AxSpA, published classification criteria designed for both populations of AxSpA.9-11 Today, many rheumatologists believe nr-AxSpA patients could benefit from biologics, and in Europe multiple therapies are approved for the indication. However, no therapies are approved for nr-AxSpA in the United States (Table 1).
“Early diagnosis is a major unmet need. The diagnostic delay is still eight years. We use questions to make the diagnosis that were basically defined 40 years ago but many people don’t have typical presentations, and I think we have to start thinking about a better set of questions to find out what is happening in those people that we’re not diagnosing early.” – Rheumatologist, United Kingdom
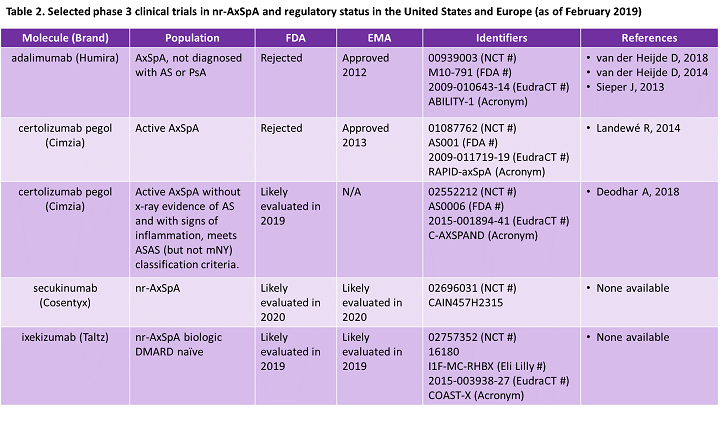
In 2019, we expect UCB’s Cimzia and Eli Lilly’s Taltz will be evaluated by the U.S. FDA for nr-AxSpA, followed by Novartis’ Cosentyx in 2020. According to Decision Resources Group (DRG), recognition of nr-AxSpA in the United States is expected to drive growth of AxSpA, with U.S. nr-AxSpA market sales projected to triple by 2027.12 Therefore, we decided to look back at the first European and U.S. regulatory evaluations of biologics for nr-AxSpA to understand potential regulatory concerns and future market dynamics.
Regional Differences In The Approval Of Biologics For nr-AxSpA: The European Perspective
Humira (AbbVie) was the first biologic approved in Europe for nr-AxSpA in 2012 (Table 1).13 In an attempt to understand the current medical understanding of this potential new indication and outstanding clinical questions, the European Medicines Agency/Committee for Medicinal Products for Human Use (EMA/CHMP) Humira assessment report states that the CHMP brought in experts to discuss the “reliability, sensitivity, specificity, and predictive value of the ASAS classification criteria,” with a focus on understanding the definition of the nr-AxSpA subgroup.14 The CHMP also sought expert opinion on current clinical understanding of and important treatment goals for AxSpA. Despite some concerns, including not knowing which nr-AxSpA patients will go on to develop AS, the CHMP recognized the unmet need for nr-AxSpA patients unresponsive to NSAIDs. Ultimately, the EMA approved label for use of Humira for nr-AxSpA specified that patients who could benefit most were those who had both clinical symptoms of AxSpA and “objective evidence of inflammation” as measured by a positive MRI or elevated C-reactive protein (CRP) that could not be otherwise explained. This restriction on the indication label was in response to concerns about the potential for misdiagnosis of patients with non-inflammatory back pain.14
Closely following Humira’s approval, Cimzia became the second biologic therapy approved for nr-AxSpA in 2013.15 Although European regulators expressed concerns about misclassification of the patient population in the original clinical trial subgroups, regulators decided that post-hoc analyses performed to address the discrepancies did not change the initial interpretation of the data. The lack of safety data for long-term use in AxSpA patients was also a concern; however, regulators did not view it as sufficient to prevent approval. Cimzia’s indication wording, like Humira’s, included “objective signs of inflammation” to address concerns about misdiagnosis.
Both Enbrel (in 2014) and Simponi (in 2015) were approved for nr-AxSpA in Europe. In October 2017, the EMA published updated guidelines for developing therapies for AxSpA, a revision of guidelines for the treatment of AS published in 2009.16 These new guidelines address the evolving understanding of AxSpA and discuss approved therapies, including IL-17 inhibitors, a relatively new class first approved for AS in Europe in 2015. Guidance on clinical trial design and preferred primary endpoints, among other things, is no doubt helpful, both to drug developers and regulators as well as to rheumatologists who make prescribing decisions. Regulatory recognition of nr-AxSpA is mirrored by interviewed European rheumatologists who widely treat AS and nr-AxSpA similarly, with one U.K. physician stating, “non-radiographic has been rather an obsolete term for us for the last 25 years.”
Regional Differences In The Approval Of Biologics For nr-AxSpA: The American Perspective
Regulatory recognition of nr-AxSpA has played out quite differently in the United States, where no biologics are approved. However, interviewed U.S. thought leaders’ perceptions of AxSpA are often similar to their European colleagues, with one U.S. rheumatologist stating:
“I prefer using ‘AxSpA.’ Basically, I think that AS and nr-AxSpA are the same disease. They’re just different classifications. Non-radiographic axial SpA does not have radiographic evidence of sacroiliitis, so we have to look for sacroiliitis on an MRI to diagnose it. But other than that, I don’t see any differentiation between the two categories. It’s a regulatory issue in this country because the FDA refuses to grant that labeling, but we’ve had long discussions about it, we’ve sent them articles and references that actually said that there are classification differences, but it’s the same spectrum of disease.” – Rheumatologist, United States
The FDA last evaluated therapies for nr-AxSpA in 2013 in a two-day meeting of the Arthritis Advisory Committee (AAC), a panel of experts who make recommendations to the FDA.17 The first day of the meeting focused on the use of ASAS criteria in diagnosing AxSpA and the current medical understanding of nr-AxSpA. An industry working group (including AbbVie, UCB, Celgene, and Pfizer) and members of the National Institutes of Health (NIH) and the Center for Drug Evaluation and Research (CDER) presented data at the meeting.
The majority of the AAC viewed the available data as insufficient to implement AxSpA as an indication. The AAC noted the lack of understanding of the natural history of the indication, particularly the inability to identify which nr-AxSpA patients would progress to AS. In addition, patient populations identified using the ASAS criteria were not as homogeneous as those identified using the mNY criteria. Members of the committee viewed the potential for inappropriate treatment with TNF-α inhibitors due to misdiagnosis of non-inflammatory lower back pain as a substantial risk. Although the AAC had similar concerns as those discussed by European regulators, the meeting summary suggests that American physicians and regulators had a more conservative interpretation of the available data and were more skeptical of a positive risk/benefit profile for biologic therapy use.
During the second day of the meeting, both Humira and Cimzia applications for approval were discussed. Humira was evaluated as a therapy for nr-AxSpA.18 Again, discussion focused on the use of ASAS criteria to define AxSpA, and members of the AAC expressed concerns about Humira’s clinical trial population, which made it difficult to evaluate the treatment effects in the nr-AxSpA group. Ultimately, 12 of 14 members voted not to recommend Humira’s approval due to the lack of sufficient efficacy data in nr-AxSpA patients.
Cimzia was evaluated as a therapy for both AS and nr-AxSpA and similar issues to those brought up for Humira were discussed, but the AAC cited three general issues that made the study insufficient to support a novel indication: (1) the nr-AxSpA population being enriched with AS patients, (2) the limited study duration (considering the potential for life-long treatment with a biologic), and (3) the magnitude of the effect in the nr-AxSpA population. Again, AAC members expressed concern about the risk to patients given inappropriate treatment, specifically citing the potential for misidentification of patients in real-world practice settings. Ultimately, seven members voted to recommend approval, with six voting no (and one abstention). However, individual members of the AAC did find the data compelling, with one saying:
“I think we saw for just about every endpoint examined there was evidence of efficacy, primary and secondary, and a priori and post hoc. So, I don’t think the primary question here is efficacy…With that said, I would be more reassured with an adequately powered prospective trial that was limited to the radiographically-negative population, based on central reads.” – Dr. Caleb Alexander, Arthritis Advisory Committee meeting, July 23, 2013
In October 2013, the FDA approved Cimzia for AS.19 However, neither Humira nor Cimzia was approved for nr-AxSpA. Interestingly, American and European regulators shared similar concerns about the use of ASAS classification criteria, the definition of the clinical trial patient populations, and the potential for misclassification of patients with non-inflammatory lower back pain. However, European regulators judged the data strong enough to warrant approval, while American regulators remained unconvinced.
The FDA meeting in 2013 appears to be the last formal public discussion about AxSpA. This lack of formal recognition likely makes it more difficult for pharmaceutical companies and government regulators when discussing development of potential therapies for the indication. However, comments made during the two-day AAC meeting do provide clues on important characteristics necessary to achieve FDA approval, including the following summary guidance on developing a sufficient clinical trial plan:
“…a [clinical] trial with multiple arms that better defines the subpopulation and more closely matches the expanded indication…CRP and MRI should be included in defining this group of patients with clarification that MRI of sacroiliac joints rather than spinal MRI should be specified and other reason for CRP elevation should be excluded.” – Summary Minutes of the Arthritis Advisory Committee meeting, July 23, 2013
Why The Difference? Then And Now
Why is there a regional difference in the acceptance of AxSpA? Although there is no one reason why European physicians and regulators have embraced AxSpA and nr-AxSpA more quickly, the initial study that developed the 2009 ASAS criteria evaluated a group of patients in Germany suggesting the unmet need of patients with nr-AxSpA has, historically, been more widely recognized by European physicians.9,10 European and American regulators have disagreed on the recognition of other indications, such as in the case of fibromyalgia, for which three drugs are approved in the United States but none in Europe.
However, in nr-AxSpA, the FDA and EMA expressed similar concerns when evaluating the same biologics but ultimately came to different conclusions. For example, although both regulatory agencies noted UCB’s post-hoc subgroup analysis based on central X-ray readings, the EMA reported the post-hoc analysis did not change the overall interpretation, while the FDA focused on the difference in the magnitude of the treatment effect between AS and nr-AxSpA groups. Ultimately, it seems that the EMA was looking at the potential benefits of approval, while the FDA was more focused on possible risks, a difference that may be in keeping with the historical founding of each organization.20 The FDA was originally formed as a centralized consumer protection agency, 21 while the EMA was founded to standardize commercial rules across the European Union. 22
An example of the FDA’s more conservative stance on risk is Novartis’s Arcapta Neohaler. First approved in Europe as Onbrez Breezhaler in 2009, it was only approved in the United States in March 2011 after additional studies supporting its safety.23,24 More recently, in 2017, the FDA gave Eli Lilly a complete response letter (CRL) for its Jak inhibitor, Olumiant, due to safety concerns and in 2018 approved only a 2-mg dose (both 2- and 4-mg doses were approved in Europe).25 Ultimately, it is unsurprising that neither Humira nor Cimzia was approved for nr-AxSpA given the lack of two “adequate and well-controlled studies that demonstrate statistical and clinically meaningful results,” the description of the ideal clinical trials required for FDA approval.
However, hope for more regulatory clarity in the United States is on the horizon. At the 2018 American College of Rheumatology/Association of Rheumatology Health Professionals (ACR/AHRP) meeting, ACR, the Spondylitis Association of America (SAA), and the SPondyloArthritis Research and Treatment Network (SPARTAN) presented draft recommendations for the treatment of both AS and nr-AxSpA (previous recommendations have included guidelines for nr-AxSpA).26,27 Suspiciously well-timed for the expected 2019 FDA evaluation of biologic applications for Cimzia (for nr-AxSpA) and Taltz (for AS and nr-AxSpA), the recommendations included both IL-17 inhibitor and Jak inhibitor drug classes in addition to TNF-α inhibitors.
“I was surprised about the different prescription behaviors between some cohorts or colleagues in the U.S. versus Europe. In terms of the new ACR recommendations, I think there are not substantial differences compared to how EULAR is recommending the treatment of AxSpA.” – Rheumatologist, Germany about ACR/SAA/SPARTAN draft guidelines presented at 2018 ACR meeting
Going Forward: What To Expect In 2019 And 2020
Three biologics are in Phase 3 clinical trials for nr-AxSpA and one (Cimzia) has reported positive top-line data (Table 2). Cimzia’s second clinical trial for nr-AxSpA, initiated after the FDA refused approval in 2013, was designed following FDA recommendations. No doubt the marketers of both IL-17 inhibitors pursuing FDA (and EMA) approval for nr-AxSpA, Taltz and Cosentyx, have worked closely with regulatory agencies to address concerns expressed in 2013. Marketers developing potential therapies for AxSpA or similar indications can learn lessons from Humira’s and Cimzia’s 2013 failures. In addition to proven efficacy and safety data, companies seeking approval in newer indications should work closely with regulators to ensure that clinical trials have a well-defined patient population and that diagnostic criteria are descriptive and quantitative enough for practical real-world use. Doing so will help to avoid concerns about misdiagnosis.
Although we expect the FDA approval of therapies for nr-AxSpA to be an important event in the coming year, questions remain about the market potential. Because three therapies are expected to be approved within a similar time frame, any first-to-market advantage may be limited, making physician experience and perception of efficacy and safety more important. In addition, access and reimbursement restrictions, including prior authorization and step therapy requirements, will likely have a large effect on uptake of these therapies. One unexpected market driver may be UCB’s extensive studies showing that Cimzia does not cross the placenta and is safe to use during pregnancy. These data may positively differentiate Cimzia from other approved therapies, especially because nr-AxSpA patients are more likely to be female and within childbearing age.28
Interviewed U.S. rheumatologists report that they currently formally diagnose nr-AxSpA patients with AS to gain access to biologic therapies. Therefore, some physicians have speculated that the FDA approval of therapies for nr-AxSpA will not change the landscape of AxSpA in the United States. However, others have made compelling arguments to the contrary:
There’s a growing understanding among U.S. rheumatologists about non-radiographic axial SpA. What we’ve been doing in recent years is calling our patients ankylosing spondylitis so that we can prescribe TNF inhibitors or IL-17 inhibitors. So, you might say “So what?” about the importance of the Cimzia clinical trial. The importance is that the FDA will have to approve Cimzia because UCB followed to the letter the request of the FDA for everything they wanted: a 52-week placebo-controlled trial, central reading of the imaging studies, etc. In my opinion, it would be virtually impossible for the agency to go back on its word. It is very clear-cut that the Cimzia-treated patients did much better than the placebo patients. The importance of approval is that now UCB and, then soon to follow, other companies will be able to go out and give promotional talks, showing these results. They’ll be able to go to educational sessions with non-rheumatologists - orthopedists, physiatrists, primary care docs, pain specialists, and teach them about the full spectrum of AxSpA. Many of these patients, especially the female patients, have been languishing in these practices without getting appropriate sacroiliac joint MRI scans, CRP values, or HLA-B27 gene markers to identify them. – Rheumatologist, United States
Based on our primary market research, DRG projects a boost in the diagnosis and treatment rates of nr-AxSpA in the United States as part of physicians’ increased awareness of the indication.12
Some interviewed rheumatologists have suggested that Cimzia’s approval will mean other TNF-α inhibitors will seek approval. However, marketers of Humira (AbbVie), Enbrel (Amgen), Remicade, and Simponi/Simponi Aria (both Janssen) will have to weigh the cost/benefit of running a clinical trial, especially considering the entry of biosimilars. In 2018, we estimated that TNF-α inhibitors accounted for almost 90 percent of the sales of biologics for the entire AxSpA market; however, we expect TNF-α inhibitor sales to fall to just over 50 percent by 2027 as rheumatologists gain more experience with IL-17 inhibitors.12 This decline may be even steeper for the nr-AxSpA subpopulation, especially if Cimzia is the only approved TNF-α inhibitor for nr-AxSpA. Whatever happens, we expect that the next few years will be exciting for rheumatologists and their patients with AxSpA as the potential of this new market segment in the United States is realized.
References:
- Baraliakos, X. & Braun, J. Non-radiographic axial spondyloarthritis and ankylosing spondylitis: what are the similarities and differences? RMD Open 4 (2014).
- Deodhar, A. Rheumatologists Make Progress Defining Spectrum of Axial Spondyloarthritis. The Rheumatologist. Available at: https://www.the-rheumatologist.org/article/rheumatologists-make-progress-defining-spectrum-of-axial-spondyloarthritis/3/. (Accessed: 18th January 2019)
- Taurog, J. D., Chhabra, A. & Colbert, R. A. Ankylosing Spondylitis and Axial Spondyloarthritis. N. Engl. J. Med. 374, 2563–2574 (2016).
- Rudwaleit, M., Khan, M. A. & Sieper, J. The challenge of diagnosis and classification in early ankylosing spondylitis: Do we need new criteria? Arthritis Rheum. 52, 1000–1008 (2005).
- Danve, A. & Deodhar, A. Axial spondyloarthritis in the USA: diagnostic challenges and missed opportunities. Clin. Rheumatol. (2018). doi:10.1007/s10067-018-4397-3
- Gossec, L. Diagnostic Delay and Associated Factors in Axial Spondyloarthritis across Europe. Results from the European Map of Axial Spondyloarthritis Survey. (2018).
- Gensler, L. Axial Spondyloarthritis Mimics. ACR - 2018 ACR/ARHP Annual Meeting Available at: https://acr.confex.com/acr/2018/meetingapp.cgi/Paper/69813. (Accessed: 18th January 2019)
- Low Back Pain Fact Sheet | National Institute of Neurological Disorders and Stroke. Available at: https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Low-Back-Pain-Fact-Sheet. (Accessed: 18th January 2019)
- Rudwaleit, M. et al. The development of Assessment of SpondyloArthritis international Society classification criteria for axial spondyloarthritis (part II): validation and final selection. Ann. Rheum. Dis. 68, 777–783 (2009).
- Rudwaleit, M. et al. The development of Assessment of SpondyloArthritis international Society classification criteria for axial spondyloarthritis (part I): classification of paper patients by expert opinion including uncertainty appraisal. Ann. Rheum. Dis. 68, 770–776 (2009).
- Sieper, J. et al. The Assessment of SpondyloArthritis international Society (ASAS) handbook: a guide to assess spondyloarthritis. Ann. Rheum. Dis. 68, ii1–ii44 (2009).
- Genco, M. et al. Axial Spondyloarthritis Disease Landscape and Forecast. Decision Resources Group. (2018).
- Abbott’s HUMIRA® (adalimumab) Approved in Europe for Treatment of Non-radiographic Axial Spondyloarthritis (nr-axSpA). Abbott MediaRoom Available at: https://abbott.mediaroom.com/2012-07-30-Abbotts-HUMIRA-adalimumab-Approved-in-Europe-for-Treatment-of-Non-radiographic-Axial-Spondyloarthritis-nr-axSpA. (Accessed: 18th January 2019)
- Humira | European Medicines Agency. Available at: https://www.ema.europa.eu/en/medicines/human/EPAR/humira. (Accessed: 18th January 2019)
- Cimzia | European Medicines Agency. Available at: https://www.ema.europa.eu/en/medicines/human/EPAR/cimzia-0. (Accessed: 18th January 2019)
- Guideline on the Clinical Investigation of Medicinal Products for the Treatment of Axial Spondyloarthritis. Available at: https://www.ema.europa.eu/documents/scientific-guideline/guideline-clinical-investigation-medicinal-products-treatment-axial-spondyloarthritis-revision-1_en.pdf. (Accessed: 25th February 2019)
- Advisory Committee Calendar > July 22, 2013: Arthritis Advisory Committee Meeting Announcement. Available at: https://wayback.archive-it.org/7993/20170404145528/https:/www.fda.gov/AdvisoryCommittees/Calendar/ucm354580.htm. (Accessed: 18th January 2019)
- Advisory Committee Calendar > July 23, 2013: Arthritis Advisory Committee Meeting Announcement. Available at: https://wayback.archive-it.org/7993/20170404145509/https:/www.fda.gov/AdvisoryCommittees/Calendar/ucm355358.htm. (Accessed: 18th January 2019)
- Press Releases | UCB. Available at: https://www.ucb.com/stories-media/press-releases/article/Cimzia-certolizumab-pegol-Approved-by-FDA-for-Treatment-of-Adults-with-Active-Ankylosing-Spondylitis. (Accessed: 21st January 2019)
- Van Norman, G. A. Drugs and Devices: Comparison of European and U.S. Approval Processes. JACC Basic Transl. Sci. 1, 399–412 (2016).
- Commissioner, O. of the. The History of FDA’s Fight for Consumer Protection and Public Health. Available at: https://www.fda.gov/AboutFDA/History/default.htm. (Accessed: 21st January 2019)
- History of EMA | European Medicines Agency. Available at: https://www.ema.europa.eu/en/about-us/history-ema. (Accessed: 21st January 2019)
- Novartis receives FDA approval for Arcapta Neohaler, a novel once-daily bronchodilator for COPD | FierceBiotech. Available at: https://www.fiercebiotech.com/biotech/novartis-receives-fda-approval-for-arcapta-neohaler-a-novel-once-daily-bronchodilator-for. (Accessed: 21st January 2019)
- Novartis launches once-daily COPD drug in US. PMLive (2012). Available at: http://www.pmlive.com/pharma_news/novartis_launches_copd_drug_us_indacaterol_394861. (Accessed: 21st January 2019)
- Genco, M. JAK inhibitor safety is front and center during regulatory approval – Drug Watch. Decision Resources Group (2018). Available at: https://decisionresourcesgroup.com/drg-blog/drug-watch/jak-inhibitor-safety-front-center-regulatory-approval/. (Accessed: 21st January 2019)
- Ward, M. ACR SAA SPARTAN Guideline Update. ACR - 2018 ACR/ARHP Annual Meeting Available at: https://acr.confex.com/acr/2018/meetingapp.cgi/Paper/69333. (Accessed: 18th January 2019)
- Ward, M. M. et al. American College of Rheumatology/Spondylitis Association of America/Spondyloarthritis Research and Treatment Network 2015 Recommendations for the Treatment of Ankylosing Spondylitis and Nonradiographic Axial Spondyloarthritis: ACR/SAA/Spartan Treatment Recommendations in as. Arthritis Rheumatol. 68, 282–298 (2016).
- Certolizumab Pegol (Cimzia®). MotherToBaby. Available at: https://mothertobaby.org/fact-sheets-parent/. (Accessed: 25th February 2019)
Table 2 References:
- Deodhar A, et. Al. Efficacy and Safety Outcomes in Patients with Non-Radiographic Axial Spondyloarthritis Treated with Certolizumab Pegol: Results from the First 52-Week Randomized Placebo-Controlled Study (NCT02552212). ACR 2018. Abstract # 1868 https://acrabstracts.org/abstract/efficacy-and-safety-outcomes-in-patients-with-non-radiographic-axial-spondyloarthritis-treated-with-certolizumab-pegol-results-from-the-first-52-week-randomized-placebo-controlled-study-nct02552212/. (Accessed 28th December 2018)
- Landewé R, et al. Efficacy of certolizumab pegol on signs and symptoms of axial spondyloarthritis including ankylosing spondylitis: 24-week results of a double-blind randomised placebo-controlled Phase 3 study. Ann Rheum Dis. 2014 Jan;73(1):39-47. doi: 10.1136/annrheumdis-2013-204231. Epub 2013 Sep 6.
- van der Heijde D, et al. Clinical and MRI remission in patients with nonradiographic axial spondyloarthritis who received long-term open-label adalimumab treatment: 3-year results of the ABILITY-1 trial. Arthritis Res Ther. 2018 Mar 27;20(1):61. doi: 10.1186/s13075-018-1556-5.
- van der Heijde D, et al. Spinal inflammation in the absence of sacroiliac joint inflammation on magnetic resonance imaging in patients with active nonradiographic axial spondyloarthritis. Arthritis Rheumatol. 2014 Mar;66(3):667-73. doi: 10.1002/art.38283.
- Sieper J, et al. Efficacy and safety of adalimumab in patients with non-radiographic axial spondyloarthritis: results of a randomised placebo-controlled trial (ABILITY-1). Ann Rheum Dis. 2013 Jun;72(6):815-22. doi: 10.1136/annrheumdis-2012-201766. Epub 2012 Jul 7.
- Eli Lilly and Company Press Release. Lilly Announces Positive Top-Line Phase 3 Results for Taltz® (ixekizumab) in Ankylosing Spondylitis (Radiographic Axial Spondyloarthritis). 13 February 2018. https://www.prnewswire.com/news-releases/lilly-announces-positive-top-line-phase-3-results-for-taltz-ixekizumab-in-ankylosing-spondylitis-radiographic-axial-spondyloarthritis-300597608.html. (Accessed: 31st December 2018).
About the Author:
 Maria Genco, Ph.D., is a business insights analyst at Decision Resources Group (DRG), specializing in pharmaceutical market analysis of immune system disorders with expertise in rheumatoid arthritis, axial spondyloarthritis, and ulcerative colitis. She holds a doctorate in neuroscience from Brandeis University, where she studied intrinsic properties of single cell neurons using Drosophila melanogaster.
Maria Genco, Ph.D., is a business insights analyst at Decision Resources Group (DRG), specializing in pharmaceutical market analysis of immune system disorders with expertise in rheumatoid arthritis, axial spondyloarthritis, and ulcerative colitis. She holds a doctorate in neuroscience from Brandeis University, where she studied intrinsic properties of single cell neurons using Drosophila melanogaster.