
Harnessing Analytics To Minimize Antibody Variants And Speed Up Development
By Yi Yang, Ph.D., Genentech

Therapeutic antibody drugs have proven to be an important type of therapeutic in treating a wide range of diseases and have shown considerable success over the past few decades. However, to date, the development process remains a costly and time-consuming effort.1 One contributing factor to the high cost and long development timeline is the establishment of a robust manufacturing process that ensures the safety and efficacy of antibody drugs. Bioprocess development involves several key steps, including the selection of a suitable cell clone, optimization of cell culture conditions to achieve high titer and desired product quality, development of purification processes to refine product quality and remove impurities, and formulation of antibodies for shelf life and long-term supply.
Throughout bioprocess development, analytical characterization plays a central and indispensable role for various activities. For example, analytical characterization of therapeutic antibody quality attributes is often the first step to identify critical quality attributes (CQAs) and to establish quality target product profiles, which help guide the development effort of antibody drugs with desired product quality to ensure product safety and efficacy. During cell culture and purification process development, analytical characterization provides critical information on product variant and impurity profiles, which supports various process optimization efforts to achieve the target product quality and to identify critical process parameters (CPPs) that lead to a robust manufacturing process. During formulation development, analytical characterization helps assess the stability of therapeutic antibodies under various storage and stress conditions. By studying quality attributes under these conditions, and investigating potential degradation pathways, aggregation, and other stability-indicating parameters, analytical characterization enables the determination of appropriate formulations to ensure stability of the antibody drugs over the product shelf life.
Additionally, analytical characterization is needed for comparability studies when changing the manufacturing process or comparing different batches or formulations of a therapeutic antibody drug. The comparability studies assess the similarity of the drug substance or drug product before and after process changes with regard to product quality, safety, and efficacy. The analytical characterization results are important for the developers to establish the comparability of therapeutic antibody drugs and to ensure that any differences are within an acceptable range and do not have negative impact on product quality, safety, or efficacy. Overall, analytical characterization is vital for assessing the quality, optimizing the manufacturing process, evaluating stability, ensuring comparability, and meeting regulatory requirements throughout the development of therapeutic antibody drugs. It enables developers to understand and control critical quality attributes, leading to the production of safe and effective antibody-based treatments.
Multiple Sources Of Therapeutic Antibody Product Variants
Therapeutic antibody drugs are inherently heterogeneous due to their large molecular size and the diverse mechanisms to form product variants during manufacturing and storage.2 For example, exposure to high temperature could result in the formation of size variants like fragments or aggregates. Post-translational modifications or chemical degradations can alter antibody charge profile and generate various charge variants. Furthermore, alterations in the protein sequence during cell culture process can lead to sequence variants or variations in the N- and C-terminal sequences.
Commonly observed fragments (or low molecular weight size variants) of therapeutic antibodies, such as Fab and desFab, are related to fragmentation in the antibody hinge region. Extensive research has identified non-enzymatic hydrolysis as the primary process responsible for hinge fragmentation in IgG1 antibody drugs.3 Characterization studies have revealed specific cleavage sites in the upper hinge region, such as between heavy chain residues Asp211 and Lys212, and between His224 and Thr225. Factors such as hinge sequence, solution pH, temperature, and excipient components can influence the extent of hinge hydrolysis and the generation of these size variants.
High molecular weight size variants, known as antibody aggregates, have also been extensively studied. These variants can range in size and structure, from small oligomers to larger particulate forms. Aggregate formation involves multiple factors, including protein-protein interactions, conformational changes, and exposure of hydrophobic regions. Conditions such as pH, temperature, and shear stress can promote intermolecular interactions among antibodies, leading to aggregate formation. Specific antibody sequences and post-translational modifications can further influence aggregate propensity. Different types of antibody aggregates may have varying immunogenic potentials, with larger aggregates posing a higher risk due to the presence of cryptic epitopes that can trigger immune responses. Thorough characterization and control of antibody aggregates throughout development and manufacturing processes are essential to ensure the safety and efficacy of antibody-based drugs.
The Complexity Of Antibody Charge Profiles
Charge variants are another common type of product variant observed in therapeutic antibody drugs. These variants result from alterations in the charge profiles of antibodies. For example, the conversion of specific asparagine residues to aspartic acid or isoaspartic acid increases the overall negative charge, leading to the formation of acidic variants. In addition, glycation of lysine and antibody N-termini can contribute to the acidic charge heterogeneity due to the formation of ketoamine Amadori products between the primary amines and reducing sugars (such as glucose). Incomplete removal of the C-terminal lysine residue during the cell culture process can elevate the overall negative charge, resulting in the formation of basic variants. The complexity of antibody charge profiles is exemplified by the identification of more than 30 distinct types of charge variants in the literature.4 Understanding and controlling the charge variant profile pose significant challenges in ensuring the desired charge variant profile and consistent product quality.
The extensive heterogeneity observed in therapeutic antibodies, encompassing size variants, charge variants, and post-translational modifications, necessitates comprehensive characterization and control strategies. By addressing the challenges posed by product variants, including their characterization and control, manufacturers can ensure the production of safe, effective, and high-quality therapeutic antibody drugs for patients.
Size Variants Identified In An IgG1 Antibody, mAb1
To illustrate the intricate nature of product variants in therapeutic antibody drugs, extensive characterization was performed on a mAb1, an IgG1 antibody, as a representative example. The characterization initially focused on molecular size heterogeneity using size-exclusion chromatography (SEC), as demonstrated in Figure 1. In the high molecular weight forms (HMWF) region, a single prominent peak was observed, while the low molecular weight forms (LMWF) region displayed two minor peaks. The LMWF peaks were isolated and subjected to mass spectrometry analysis. As depicted in Figure 2, the deconvoluted mass spectra of intact LMWF2 revealed that LMWF2 primarily consisted of various forms of Fab fragments resulting from cleavages in the upper hinge region. Specifically, the major mass species were identified as cleavages between heavy chain residues Asp211 and Lys212, as well as between His224 and Thr225, consistent with previously reported common fragments in IgG1.3 For LMWF1, the deconvoluted mass spectra of intact LMWF1 (Figure 3) indicated that it mainly consisted of Fc fragments resulting from hinge cleavages between heavy chain residues Thr225 and Cys226 and between Pro227 and Pro228. The distinct cleavage sites suggest that the formation mechanism of LMWF1 is likely different from that (non-enzymatic hydrolysis) observed in the upper hinge region for the Fab fragments. It should also be noted that, as a result of the observed hinge cleavages, the corresponding desFab and F(ab’)2 fragments likely co-eluted with the main peak. The SEC method may not have the resolution to completely separate these size variants from the main peak. In certain instances, these size variants have been observed as a main peak shoulder on the size exclusion chromatograms.
In addition, the HMWF peak was fractionated and characterized by SEC-multiangle light scattering (SEC-MALS). The analysis revealed that the HMWF fraction predominantly consisted of dimers (2.97 x 105 g/mol, Rh=7.3 nm) compared to the monomer form (1.47 x 105 g/mol, Rh=5.5 nm). Subsequently, the HMWF fraction was analyzed by CE-SDS (data not shown), which showed that 65% of the aggregates remained non-dissociated under non-reducing conditions and indicates that these aggregates are likely covalent dimers. Following deduction with dithiothreitol (DTT), the majority of the aggregates converted into light chain and heavy chain, with only 11% remaining, likely originating from non-reducible aggregates or incomplete reduction.
In summary, the characterization of mAb1 revealed size variants in the form of LMWF and HMWF. The LMWF consisted of various forms of Fab and Fc fragments, while the HMWF primarily comprised covalent (likely linked via disulfide bond) dimers.
Figure 1 Analysis of mAb1 Molecular Size Distribution by SEC
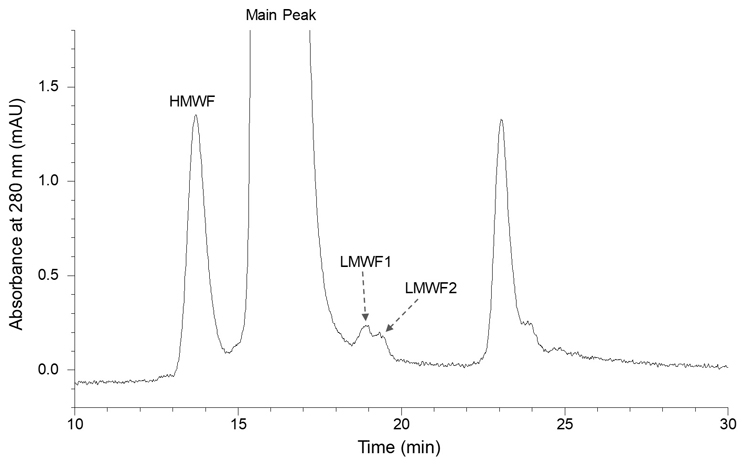
HMWF = high molecular weight forms; LMWF = low molecular weight forms.
Figure 2 Deconvoluted Mass Spectra of Intact LMWF2
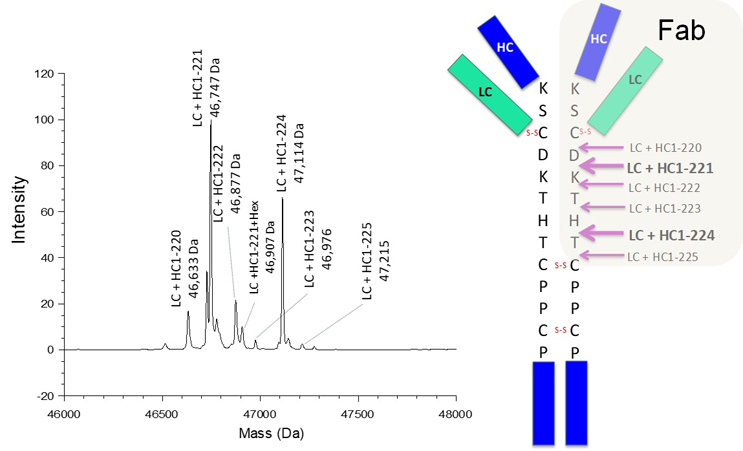
The observed masses from intact LMWF2 are consistent with the Fab portion of mAb1, which is the result of cleavages occurring in the upper hinge region (illustrated in the cartoon on the right side).
Figure 3 Deconvoluted Mass Spectra of Intact LMWF1
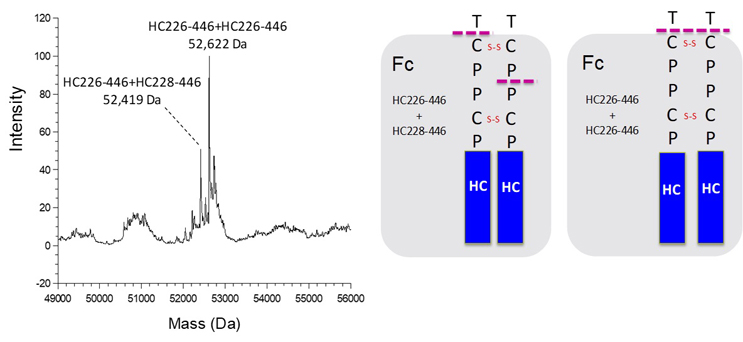
The observed masses from intact LMWF1 are consistent with the Fc portion of mAb1, which is the result of asymmetric and symmetric hinge cleavages (illustrated in the cartoon on the right side).
Charge Variants Identified In mAb1
The charge heterogeneity of mAb1 was assessed using ion exchange chromatography (IEC). Initially, mAb1 was treated with carboxypeptidase B to remove the heavy chain C-terminal lysine. Subsequently, it was separated into three regions, namely the acidic region, main peak, and basic region, as demonstrated in Figure 4. Individual peaks, labeled as AV1 to AV4, MP1, main peak, and BV1 to BV3, were fraction collected and subjected to LC-MS/MS tryptic peptide mapping. Site-specific modifications were identified based on the acquired mass spectra, and the relative levels of these modifications in each fraction were determined using extracted ion chromatograms.

Comparing the levels observed in the main peak, increased presence of deamidation, tryptophan and methionine oxidation, hydroxylysine, carboxymethyl-lysine (CML, an advanced glycation end product), N-terminal pyroglutamate, and glycation were observed in the acidic fractions (AV1 to AV4). Conversely, the basic fractions (BV1 to BV3) showed increased presence of Asp isomerization (formation of succinimide), VHS leader sequence, methionine oxidation, and proline amidation.
It is important to note that more acidic variants were detected than basic variants, indicating greater complexity in acidic charge heterogeneity. The increased complexity with acidic variants is likely related to more degradation reactions or post-translational modifications that lead to the loss of positive charge or the gain of negative charge. Furthermore, it should be noted that certain modifications, such as methionine and tryptophan oxidation, do not directly alter the net charge of the antibody molecule. The localization of these product variants is likely associated with non-charge-related interactions with the ion exchange stationary phase, as ion exchange chromatography involves mixed-mode separation mechanisms. These non-charge-related factors or interferences can be addressed through non-stationary based techniques, such as imaged capillary isoelectric focusing (iCIEF). This enables a more consistent comparison of charge profiles based on the intrinsic net charge of antibody variants. However, it should also be noted that characterization of antibody charge variants via iCIEF fractionation or by online iCIEF-MS analysis necessitates specialized instruments, and the identified charge variant distributions could differ from those identified via IEC fractionation.
Figure 4 Charge Variants Identified in mAb1
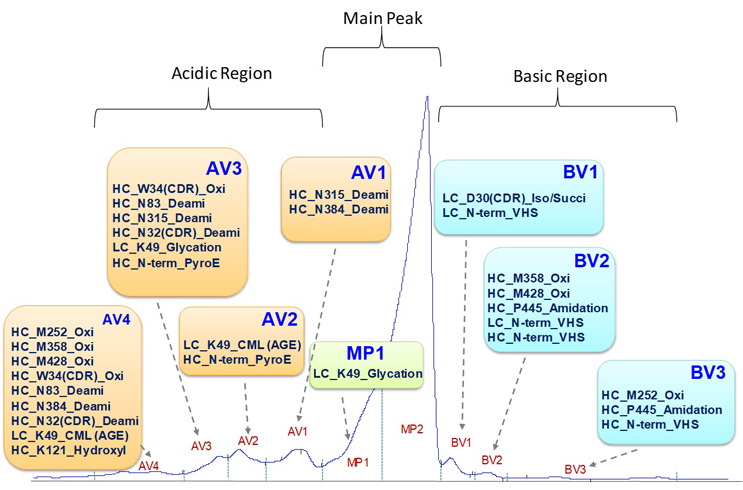
AGE = advanced glycation end product; CML = carboxymethyllysine; Deami = deamidation; Iso/Succi = isomerization/succinimide; Oxi = oxidation; PyroE = pyroglutamate
Benefits Of In-depth Product Variant Characterization For Bioprocess Development
In-depth product variant characterization provides valuable insights into the specific variants present in therapeutic antibody drugs. This information enables developers to identify critical process parameters to minimize the formation of undesirable variants. Moreover, it facilitates targeted bioprocess optimization efforts to enhance product quality and process consistency, which leads to reduced need for rework or reprocessing, ultimately improving process efficiency and reducing production costs.
For example, by characterizing mAb1 size variants, the knowledge enables a targeted effort on optimization of temperature, buffer composition, and pH throughout the manufacturing process to reduce the likelihood of upper hinge hydrolysis that generates LMWF2 (Fab). On the other hand, formation of LMWF1 (Fc) likely involves a different mechanism (yet to be discovered), which may require alternative approaches for an effective control. For HMWF (disulfide-linked dimers), optimizing pH and redox conditions during cell culture processes and reducing shear stress from mechanical processes can help minimize disulfide bond shuffling and subsequent dimer formation.
With respect to charge variants, a comprehensive characterization of these variants plays a crucial role in efficient bioprocess development to achieve the desired charge profiles, considering that diverse mechanisms are responsible for generating these variants. For mAb1, the in-depth characterization revealed that deamidation variants are a major component in the acidic region. Therefore, based on the mechanism of asparagine deamidation, mitigating acidic charge profile variation can be achieved by optimizing temperature, buffer composition, and pH during manufacturing and storage. For the basic charge profile, controlling copper concentration in cell culture media and selecting appropriate cell lines effectively manages C-terminal proline amidation variants, which result from post-translational modification by peptidylglycine alpha-amidation monooxygenase (PAM) during cell culture processes.5
It is worth noting that other product variants, such as glycosylation, thiol-related modifications, and sequence variants, also require attention. Similar to the size and charge variants, thorough understanding of these product variants leads to focused optimization efforts, which reduces the parameter space and enhances cost and time efficiency in bioprocess development.
Conclusion
The benefits of in-depth product variant characterization in antibody development are substantial. By investing in thorough product variant characterization, bioprocess development teams can streamline their efforts, focusing on key process parameters and reducing the overall complexity of process development and optimization. This knowledge-driven approach helps developers save time, reduce costs, and enhance the overall efficiency of antibody development. Ultimately, it enables a risk-based approach to process development and control strategy and consistent production of safe and effective therapeutic antibody drugs, benefiting patients and advancing the field of biopharmaceuticals.
Acknowledgement: The author would like to thank David A. Michels and Vikas Sharma for their thoughtful review of this article and Anna Mah and Fan Zhang for providing the characterization results of mAb1.
References:
- Kelley, B., Industrialization of mAb production technology: the bioprocessing industry at a crossroads. MAbs 2009, 1 (5), 443-52.
- Liu, H.; Gaza-Bulseco, G.; Faldu, D.; Chumsae, C.; Sun, J., Heterogeneity of monoclonal antibodies. J Pharm Sci 2008, 97 (7), 2426-47.
- Cordoba, A. J.; Shyong, B. J.; Breen, D.; Harris, R. J., Non-enzymatic hinge region fragmentation of antibodies in solution. J Chromatogr B Analyt Technol Biomed Life Sci 2005, 818 (2), 115-21.
- Beck, A.; Nowak, C.; Meshulam, D.; Reynolds, K.; Chen, D.; Pacardo, D. B.; Nicholls, S. B.; Carven, G. J.; Gu, Z.; Fang, J.; Wang, D.; Katiyar, A.; Xiang, T.; Liu, H., Risk-Based Control Strategies of Recombinant Monoclonal Antibody Charge Variants. Antibodies (Basel) 2022, 11 (4).
- Jiang, G.; Yu, C.; Yadav, D. B.; Hu, Z.; Amurao, A.; Duenas, E.; Wong, M.; Iverson, M.; Zheng, K.; Lam, X.; Chen, J.; Vega, R.; Ulufatu, S.; Leddy, C.; Davis, H.; Shen, A.; Wong, P. Y.; Harris, R.; Wang, Y. J.; Li, D., Evaluation of Heavy-Chain C-Terminal Deletion on Product Quality and Pharmacokinetics of Monoclonal Antibodies. J Pharm Sci 2016, 105 (7), 2066-72.
About The Author:
 Yi Yang is a senior principal scientist in the protein analytical chemistry department at Genentech where he currently serves as the head of analytical method validation and transfer. He is the analytical lead for several late-stage antibody projects, overseeing the characterization of antibody product variants and the development, validation, and transfer of analytical assays. He also is responsible for establishing comprehensive end-to-end control strategies. With over 20 years of experience in the biopharmaceutical industry, he has 12 publications and three patents. He holds a Ph.D. in pharmaceutical chemistry from the University of Kansas.
Yi Yang is a senior principal scientist in the protein analytical chemistry department at Genentech where he currently serves as the head of analytical method validation and transfer. He is the analytical lead for several late-stage antibody projects, overseeing the characterization of antibody product variants and the development, validation, and transfer of analytical assays. He also is responsible for establishing comprehensive end-to-end control strategies. With over 20 years of experience in the biopharmaceutical industry, he has 12 publications and three patents. He holds a Ph.D. in pharmaceutical chemistry from the University of Kansas.