
Interacting With FDA During Biosimilar Development: From Initial Advisory Meeting To BLA
By Mary Lou Zett, Ph.D., Regulatory Affairs, Pharmatech Associates

By Mary Lou Zett, Ph.D., Regulatory Affairs, Pharmatech Associates
In Part 1 we covered important steps in the biosimilar development path to identify and evaluate critical quality attributes for both the candidate biosimilar and the reference product, along the regulatory timeline. To further explain the process, in Part 2, we map out the milestones needed for a 351(k) Biologics License Application (BLA).
Now we will discuss what information is critical to support the sponsor’s argument that the proposed product is biosimilar to the designated reference product and when is it appropriate and at what level of detail it should be for each stage of interaction with the FDA.
When you have built a convincing argument justifying the acceptance of the proposed biosimilar, you have what you need for your first meeting with the FDA to determine if the proposed product qualifies for a 351(k) application. This first meeting is the Biosimilar Initial Advisory (BIA) Type 1 meeting.
Formal Meetings With The FDA
For your first encounter with the FDA, you should have complete analytical reports comparing the proposed biosimilar to the reference product, signed and audited. All analytical data must be traceable to the formulation intended to represent the proposed biosimilar that will be used in subsequent nonclinical and clinical assessments.
Any safety literature you can find on the reference product adds value to your position with the FDA that the proposed biosimilar is also safe. Remember, the goal of this first meeting with the FDA is to qualify the proposed biosimilar under section 351(k) of the Public Health Service (PHS) Act. You are allowed one BIA meeting and one Biosimilar Product Development (BPD) Type 4 meeting for a particular biosimilar or interchangeable product. However, you may request as many BPD Type 2 and Type 3 meetings as needed to support the development and review of a biosimilar or interchangeable product. Try to keep the number of meetings to a minimum by grouping related questions. If the proposed biosimilar has multiple indications, the meeting request should go to the division that has oversight for the reference product.
It is important to remember that a meeting request will be considered incomplete and will be denied unless the final briefing package is submitted with the request. Also, sponsors must pay a BPD fee to participate in the BPD program, which begins after the BIA. The BPD fee is a per-product annual fee, not a per-meeting or per-review fee, and the annual fee continues until the filing of a marketing application or the discontinuation of the BPD program.
Biosimilar Initial Advisory Meeting
The BIA Type 1 meeting is for determining whether or not licensure under section 351(k) of the PHS Act may be feasible in terms of demonstrating biosimilarity for the proposed product. The meeting should be designed to get feedback as to the appropriateness of the biosimilar development program as presented at this initial meeting. This is not a designation of biosimilarity but rather an evaluation of the evidence presented that it is a potential candidate.
While this early meeting with the FDA does not involve substantive review of summary data or full study reports, you must have convincing preliminary comparative analytical similarity data from at least one lot of the proposed biosimilar product compared to the U.S.-licensed reference product to present in the meeting package. The BIA is held within 90 days of receipt of the request.
Biological Product Development Meeting
The BPD Type 1 is essentially reserved for any of a few reasons, such as safety concerns that might arise during the clinical development program or resolution of disputes over FDA regulatory action(s), including an incomplete response letter or a refuse to file. These meetings are held within 30 days of receipt of the meeting request.
Make good use of the BPD Type 2 meeting. This is where you have the opportunity to discuss a specific issue (e.g., processing critical quality attributes and their rankings or any proposed chemistry, nonclinical, or clinical study design or endpoints). Use this tool to retain any ongoing targeted advice from the FDA throughout the development program. Just remember that this type of meeting does not include review of the full study report; only a substantive review of a summary may be presented. These meetings are held within 75 days of receipt of the meeting request.
There can be more than one BPD Type 3 meeting. In preparation for this type of meeting, be sure to have compiled a comprehensive analytical summary of similarity data that permits the FDA to make a preliminary evaluation of analytical similarity during development. The level of analytical data provided should be similar to what the sponsor intends to submit in a 351(k) BLA (e.g., full study reports for a clinical study or clinical studies and/or data sets that support the full study reports). Based on the data and/or data sets and results reported in the full study reports, the FDA now expects to be provided with an update on the product development plan of the proposed biosimilar, including what has been accomplished and what remains to be done. Use this opportunity to include any proposals for planned additional studies or a proposal for extrapolation. By the time you are ready for the BPD Type 3 meeting, the briefing package(s) should be well organized to serve as a template for the submission of the 351(k) BLA. These meetings are scheduled within 120 days of receipt of the meeting request.
Pre-submission: Reaching The Goal Line
A BPD Type 4 meeting is the pre-submission meeting. The purpose of this meeting is to discuss the format and content of the planned submission. Be clear as to the studies that will be used to support a demonstration of biosimilarity. Clear up any potential review issues that have surfaced during previous meetings and/or FDA feedback. Identify any ongoing studies or additional planned studies to adequately address the Pediatric Research Equity Act. This type of meeting is analogous to a Type B Pre-BLA meeting. These meetings are scheduled within 60 days of receipt of the meeting request.
The importance of this meeting in terms of a successful outcome is to make sure the FDA reviewers are acquainted with the general information to be submitted in the marketing application (including technical information), so that through discussion with the FDA the best approach for the presentation and formatting of the data in the marketing application is confirmed.
Be sure to rehearse, and in the rehearsal anticipate what questions might arise. Be sure that all previous regulatory feedback received from the FDA has been fully addressed. The following road map will take you to a successful outcome. By the time you reach the BPD Type 4 meeting, your abbreviated BLA is well on its way to approval. Opportunities to request guidance from the FDA should have been taken full advantage of by now, and final questions for the BPD Type 4 meeting should summarize the conclusion of outcomes with highlights on issues resolved throughout the review process. If further issues arise during this meeting, the final submission should address those last-minute concerns. Use the minutes from this meeting to resolve any loose ends or issues that arise.
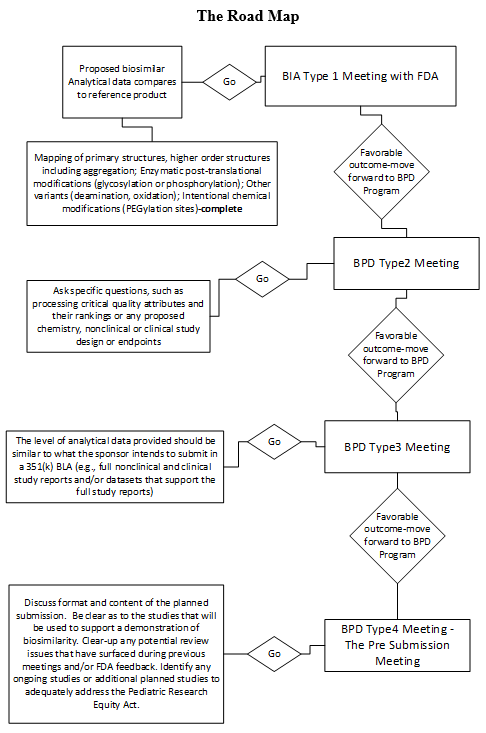
Reserve BPD Type 1 meetings for resolving any unforeseen problems that arise, such as clinical safety issues placing studies on clinical hold or disputes over decisions being discussed with the FDA. A BPD Type 1 meeting may be requested any time there is the need throughout the BPD program.
Labeling
The final approval of the biosimilar BLA is contingent upon the labeling requirement being met in Module 1 of the eCTD BLA. The FDA recommends that biosimilar product labeling incorporates relevant data and information from the reference product labeling. If a medication guide is required, applicants should develop one for the biosimilar. For patient information, use relevant information from the patient information for the reference product. Some labeling may be modified, if needed. For example, instructions for use (IFU) for the proposed biosimilar product should incorporate relevant information from the IFU for the reference product, but if the proposed biosimilar IFU differs from the reference product or images are needed to describe the biosimilar product accurately, this modification would be important to describe the biosimilar product accurately. Any such changes to labeling should be discussed with the agency. A full and final review of proposed product labeling, including the IFU, will occur as part of the 351(k) application.
When it comes to promotional labeling, such as printed, audio, or visual matter descriptive of a drug for which the labeling is disseminated by or on behalf of a drug’s manufacturer, packer, or distributor, the FDA makes recommendations, not legal requirements, that present the agency’s thinking regarding how promotional labeling should be displayed.
Given the entire process of developing, comparing, and negotiating the approval of a biosimilar, the timeline to final approval is typically eight years compared to 12 years for the reference material, and the development costs can be 10 to 20 percent of the cost of the reference material. Not only will the biosimilar pipeline be reviewed by the FDA but also the quality management system that generated traceable, trackable data sets.
Additional Resources:
- https://purplebooksearch.fda.gov/about
- Nagel, K. Introduction to Biologic and Biosimilar Product Development and Analysis, AAPS 2018.
- https://www.gabionline.net/Biosimilars
- Daller J. Biosimilars: A Consideration of the regulations in the United States and European union. Regul Toxicol Pharmacol. 2016;76:199-208.
About The Author:
 Mary Lou Zett, MBA, Ph.D., CQE, is a regulatory affairs professional with experience bringing products from research through to commercialization, encompassing small, large, biotechnology-NME and biosimilars, gene therapy, and medical device (critical, nonclinical, combination products, and in vitro diagnostics). She has prepared responses to regulatory authorities in the U.S., EU, and Canada, including electronic submissions for all types of regulatory applications and reporting, worldwide. She holds a doctorate in immunology from Rutgers University and an MBA from Fairleigh Dickinson University.
Mary Lou Zett, MBA, Ph.D., CQE, is a regulatory affairs professional with experience bringing products from research through to commercialization, encompassing small, large, biotechnology-NME and biosimilars, gene therapy, and medical device (critical, nonclinical, combination products, and in vitro diagnostics). She has prepared responses to regulatory authorities in the U.S., EU, and Canada, including electronic submissions for all types of regulatory applications and reporting, worldwide. She holds a doctorate in immunology from Rutgers University and an MBA from Fairleigh Dickinson University.