
Key Post-Pandemic Trends In Global FDA Observations For Drug Facilities
By Ajay Pazhayattil and Marzena Ingram

The FDA inspection trend insights provide powerful resources for understanding areas of regulatory focus and a benchmark for evaluating potential vulnerabilities within the quality system and beyond. The publicly available inspection observation data1 offers practical insight into mitigating many of the issues manufacturers of FDA-regulated drug products face. Organizations can use such insights2 to better understand how certain variables affect compliance and prepare for future inspections. FDA Form 483 is issued to a firm’s management at the conclusion of an inspection when an investigator(s) has observed any conditions that, in their judgment, may constitute violations of the Food, Drug, and Cosmetic (FD&C) Act and related requirements. The FDA investigators ensure that each observation noted on the FDA Form 483 is clear, specific, and significant. A comprehensive review of post-pandemic observations for drug facilities sheds light on the state of compliance and regulatory practices in the pharmaceutical industry.
We undertook a meticulous analysis of inspection data spanning 23 months, from July 1, 2021, to May 31, 2023, following the resumption of U.S. domestic inspections on July 1st, 2021.3 This extensive 483 data set from the FDA provides valuable insights into the ongoing efforts to ensure the safety and quality of pharmaceutical products. In some cases, 483 forms are manually prepared by the FDA, which are excluded from this assessment. The agency updates the data sets weekly, ensuring these findings reflect the most recent inspections and observations.
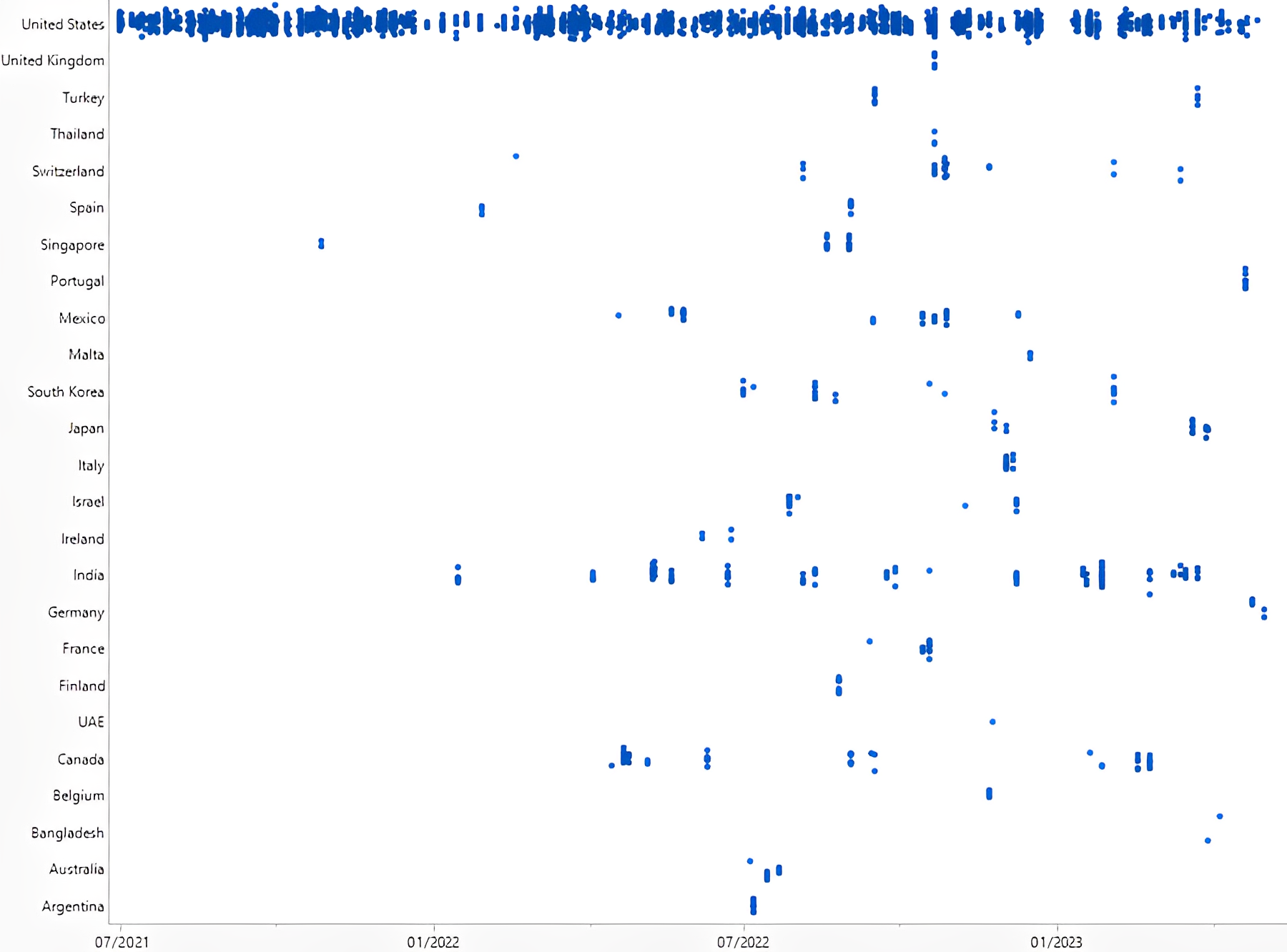
Figure 1: Observations Listed: July 1, 2021, to May 31, 2023. Click on image to enlarge.
As the world emerged from the pandemic, the FDA aimed to ensure the safety and quality of pharmaceutical products produced domestically within the United States. This is evident from the review that the inspectorate dedicated considerable attention to domestic inspections in the last two quarters of 2021. The review period encompassed numerous domestic inspections as the industry rebounded from the pandemic's impact. However, it is worth noting that foreign drug facilities also underwent scrutiny. The top 10 foreign countries with the highest FDA Form 483 observations during the last 23 months were India, Canada, Mexico, Switzerland, Singapore, South Korea, Japan, Israel, France, and Italy.
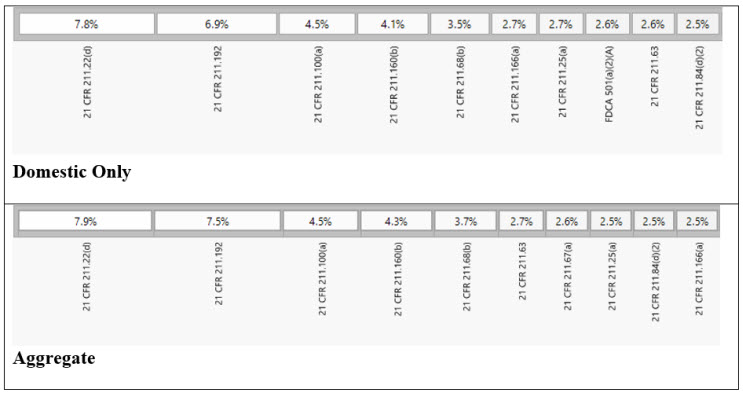
Figure 2: 21 CFR 211 Sections Observed
Among the key findings, five specific sections of the current good manufacturing practice (cGMP) regulations were the most frequently cited in the issued observations. These sections, namely 21 CFR 211.22(d), 21 CFR 211.192, 21 CFR 211.100(a), 21 CFR 211.160(b), and 21 CFR 211.68(b)4 accounted for 28% of all 483 observations during the review period. Each section addresses crucial aspects of quality control, production and process control, laboratory controls, and computer system management. For instance, 21 CFR 211.192 requires drug product production and control records, including packaging and labeling, to be reviewed and approved by the quality unit before batch release or distribution. It stresses the need for a thorough investigation of discrepancies or failures in drug product production and control, extending the investigation to related batches and products. 21 CFR 211.160(b) mandates laboratory controls to establish specifications, sampling procedures, and calibration standards for ensuring conformity of components, materials, and drug products to quality standards. The remaining top-cited sections outline requirements for written procedures, laboratory controls, and computer system controls, emphasizing the significance of adherence to specifications, standards, sampling plans, and data integrity.
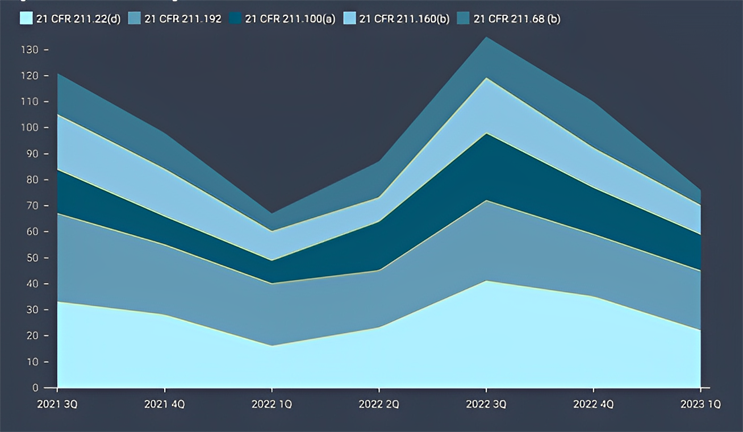
Figure 3: Trend for The Top 5 Cited Sections
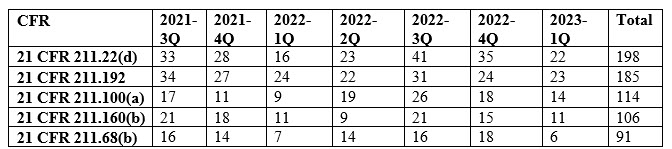
Table 1: Observation Trend for The Top 5 Cited Sections
Inspection Classification
A closer examination of the inspection end dates for 2022 and 2023 reveals distinct patterns in the number of critical observations made during different quarters. The data indicates that the first quarter showed a relatively lower number of critical observations, while the third quarter recorded the highest findings. As anticipated, with the higher number of domestic inspections, the Official Action Indicated (OAI) classification was notably higher for domestic sites. The ratio of Voluntary Action Indicated (VAI) to No Action Indicated (NAI) was, however, found to be higher for foreign sites versus domestic sites.
The final inspection outcome classifications for foreign sites, especially with OAI determination, were analyzed. More than 45% of the OAI-classified foreign sites were in India (45.9%), followed by Mexico (13.5%) and Canada (8.1%).5 Countries with 30 or more inspection classifications, i.e., India, Canada, and Germany, were selected for this analysis. Of the total facility classification determinations from India, 11.5% resulted in an OAI, while the rate was only 5.4% and 2% for Canada and Germany, respectively. The high OAI issuance rate in India is in line with what is being observed domestically (12%). This indicates the need for continued oversight of Indian manufacturing facilities.
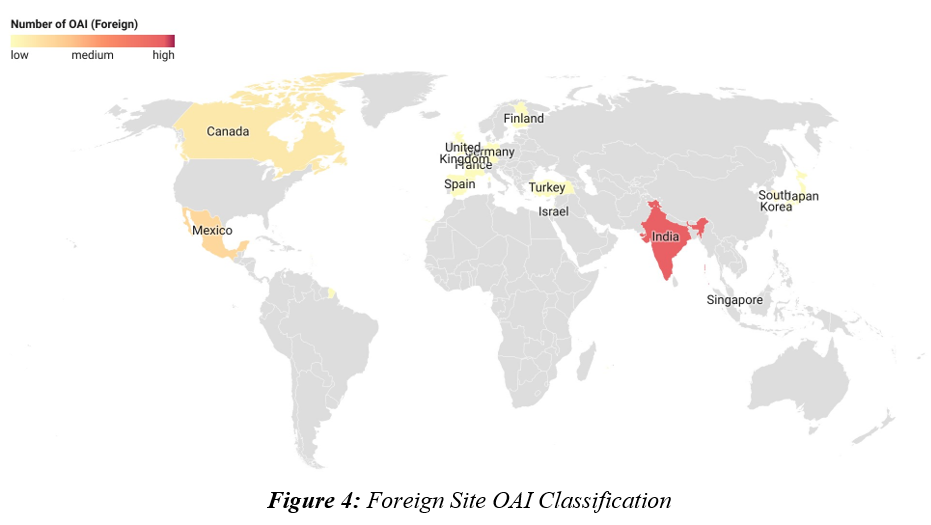
Figure 4: Foreign Site OAI Classification. Click on image to enlarge.
What Are The Correlations?
We performed a comprehensive multivariate analysis of FDA 483 observations issued to facilities in 25 countries. This analysis considered factors including total observation counts, the top five observations, and the classification of NAI, VAI, and OAI. The aim was to uncover potential relationships and correlations between these factors. Upon conducting non-parametric Spearman ρ and Kendall τ correlation analyses,6 a strong positive correlation was identified between the OAI classification and three specific observation classes: 21 CFR 211.68(b), 21 CFR 211.100(a), and 21 CFR 211.22(d).
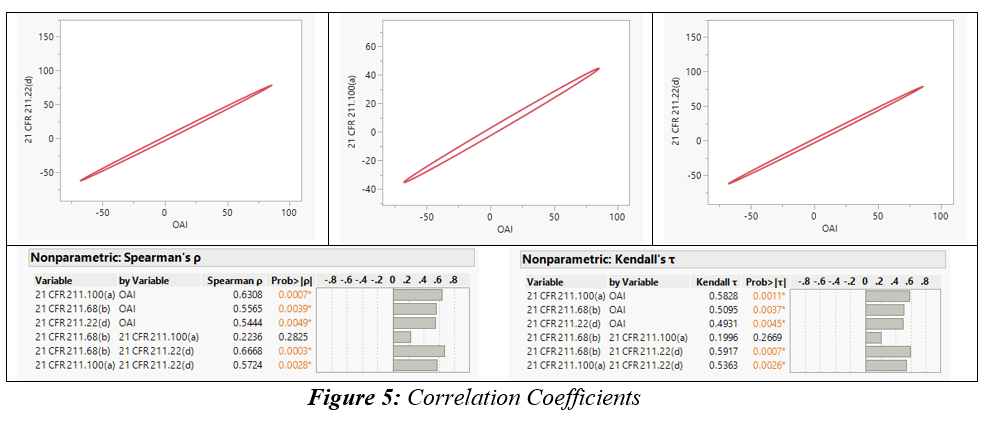
Figure 5: Correlation Coefficients. Click on image to enlarge.
21 CFR 211.68(b) emphasizes the importance of appropriate controls over computers or related systems. Authorized personnel should be responsible for making changes to master production and control records or other records, ensuring data integrity. Input and output of formulas, records, or data into the computer system must undergo accuracy checks to minimize errors. The degree and frequency of verification should be determined based on system complexity and reliability. Backup files of entered data should be maintained, except for specific automated processes. If data is eliminated, a written record of the program and validation data must be preserved. To secure backup data from alteration or loss, hard copies or alternative systems like duplicates, tapes, or microfilm should be used. Under 21 CFR 211.100(a), pharmaceutical manufacturing facilities are required to establish written procedures for production and process control. These procedures ensure that drug products maintain the intended identity, strength, quality, and purity as indicated or represented. It is essential that the drafting, reviewing, and approval of these procedures, including any subsequent changes, are carried out by the appropriate organizational units. Additionally, the quality unit should also review and approve these procedures. In accordance with 21 CFR 211.22(d), the quality control unit is required to have written procedures outlining their responsibilities and applicable procedures.
The analysis indicates that facilities with numerous observations related to a) the absence of system controls and b) the lack of written procedures for production, process, or quality control unit are more prone to receiving an OAI classification. Organizations should prioritize efforts to enhance compliance in the identified sections, thereby mitigating the risk of receiving an OAI classification during inspections. These insights should also influence the organization’s regulatory compliance-related decision-making. This assessment underscores the criticality of adhering to the cGMP regulations and serves as a reminder to consistently monitor them. Compliance in critical areas has a direct impact on regulatory outcomes. The industry should continue striving to apply the best practices, leveraging the additional insights gained from the analysis.
References
- U.S. FDA, Citations Data from Form FDA 483 Citations
- A. Pazhayattil, M. Ingram, N. Sayeed, (2020) A Quantitative Study of US FDA Inspection Data for Drug Manufacturing Sites, Therapeutic Innovation & Regulatory Science volume 54, pages 725–730
- K. Dunleavy, (2021) With Pandemic Restrictions Lifted, FDA Restarts U.S. Inspections and Seeks to Whittle its Massive Backlog, Fierce Pharma
- U.S. FDA, Code of Federal Regulations Title 21, PART 211, Current Good Manufacturing Practice for Finished Pharmaceuticals
- U.S. FDA, Inspections Classification Details
- JMP, Nonparametric Correlations Produce nonparametric measures of association between two continuous variables (Spearman’s Rho, Kendall’s Tau, and Hoeffding’s D)
 About The Authors:
About The Authors:
Ajay Babu Pazhayattil, DBA, is a management consultant at cGMP World Inc. helping clients to achieve their business goals while adhering to global regulatory compliance mandates. Previously, he was in leadership positions at a major CDMO and generic pharmaceutical organizations. He has contributed to key remediations and strategic organizational initiatives. You can connect with him on LinkedIn.
 Marzena Ingram is a senior pharmaceutical consultant with Validant. She guides clients in navigating the landscape of quality and compliance. Ingram has developed specialized teams and spearheaded the implementation of programs to meet global regulatory requirements at solid oral companies, API manufacturing sites, and 3PL organizations. You can connect with her on LinkedIn.
Marzena Ingram is a senior pharmaceutical consultant with Validant. She guides clients in navigating the landscape of quality and compliance. Ingram has developed specialized teams and spearheaded the implementation of programs to meet global regulatory requirements at solid oral companies, API manufacturing sites, and 3PL organizations. You can connect with her on LinkedIn.