
The Digital Future Of CMC Regulatory Affairs
By Raz Eliav, Beyond CMC

Drug development generates enormous amounts of data. In part 1 of this series, we appreciated the vast amounts of data that is generated in CMC development and manufacturing operations and saw how it is too much to handle in today’s document-centric systems, even if they are electronic.
The transition to FAIR (findable, accessible, interoperable, reusable) data management principles is inevitable as the data fingerprints of the advanced products of today are no longer possible to capture in documents only.
The communication of data between the life sciences industry and the authorities remains document-centric (by law), which means it is riddled with narratives, slow, and prone to bias and misinterpretation. It is imperative for both the regulators and the industry to expedite the drug approval process, especially for orphan drugs and unmet clinical needs. To do so, we must become more efficient with our data and reap its full benefits. Contrary to prior technological innovations, this time it is the regulators that are showing us the way and it is prudent that we follow their lead.
In this article, we will discuss recent initiatives from the FDA, such as KASA, PQ/CMC, and the ICH SPQS and see how they could be the early birds of the paradigm shift from electronic CTDs to digital CTDs. We will address what steps the industry should be taking in parallel with the authorities in preparation for this (very near) digital future.
Expediting Drug Time To Market
Drug development is slow as it is. Clinical development takes time and so does manufacturing process development. The regulatory approval for marketing must consider the full body of available data and is therefore, by definition, on the critical path.
ICH Q9 points out that drug shortages, especially of lifesaving ones, can translate to very immediate and clear risks to patients who rely on them. This is stated with regard to failed manufacturing batches, but this same principle can be applied to drug development as a whole. Each unnecessary delay in the development process could potentially translate to a lost life, so it is an ethical imperative to expedite the drug development and approval process, both by the regulators and the industry.
The authorities nowadays offer a number of expedited pathways, such as PRIME in Europe and Fast Track and Breakthrough designations in the U.S. Approval in these cases is based on less clinical data than usual and relies on post-marketing commitments to complete the dossier. This enables earlier access to potentially lifesaving drugs, at least for the patients at the highest risk.
The ethical tipping point of the risk-benefit balance is dictated by the underlying disease: when life is at stake, being overly cautious is unethical. But the case for expediting the process holds for non-lifesaving drugs as well. Delays due to slow authoring and review and mistakes in data analysis are unacceptable from a business point of view in a world that has artificial intelligence and advanced data sciences in it.
CTD Module 3: The Story Of CMC
The regulatory process for CMC approval revolves around Module 3 of the CTD. This document is an ICH harmonized structure (see ICH M4Q) that covers all the relevant aspects of a given product’s CMC portfolio. From the characteristics of the drug to the way it is manufactured, how its quality is controlled, and how it evolved, the entire story of CMC is told in those chapters.
The CTD structure has been codified into an electronic standard (eCTD) that is now the norm. The chapters are separate PDF documents, referenced in a common XML structure, which allows quick access and versioning.
We’ve come a long way since the days of paper submissions in trucks, but, as discussed above, these submissions remain document-centric, riddled with narrative, and intended for other humans to review.
Source: FDA presentation https://www.fda.gov/media/162865/download
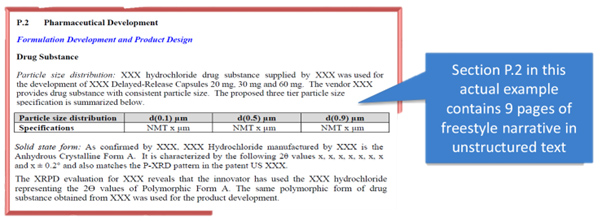
Some chapters are inevitably dense with narrative and unstructured data, like the development sections, but others present actual data tables, such as, for example, the sections outlining the full batch analysis done throughout development. These are the richest “golden troves” for data analysis in the submission, but when submitted as PDFs, they remain largely inaccessible to machines and very hard to re-analyze, interpret, and consolidate with data and narratives from other chapters.
The PQ/CMC Project
The FDA has recently published a (second) call for comments on its PQ/CMC initiative, which is looking to augment the contextual framework of the CTD. The initiative breaks down the CTD chapters into their data constituents and looks for common terminology that describes each of these constituents. The standard is developed in accordance with the Health Level 7 (HL7), Fast Health Interoperable Resources (FHIR) representation. The project is led by the HL7 Biomedical Research & Regulation (BR&R) workgroup and is open for public consultation with no deadline for comments as it is work in progress.
Each constituent of the PQ/CMC ontology is a data element that includes the data itself (which could be textual, numeric, Boolean, graphic, etc.) and, equally important, its metadata, which provides the context and interconnections with other data elements.

By breaking the CTD into a granular structure, we are taking a first step toward the ability to use a structured content and data management (SCDM) system to describe CMC and feed into automated processes and knowledge management systems such KASA, further discussed below.
The recent call for comments was to apply these foundations to an oral solid dosage form, which represents a large class of drugs and can serve as a well-understood starting point for FDA and the industry. The FDA will not stop there, of course, and the intention is to apply the PQ/CMC initiative to all types of products, as implied in the call for comments.
The FDA is not alone, and similar initiatives are forming in other regions as well. ICH is also on board with its Structured Product Quality Submissions (SPQS), though other priorities are foreseen for the revision of ICH M4Q, which is long overdue for Revision R2, as it must first address other priorities such as quality by design and advanced therapies.
KASA: A Home For Quality Knowledge
In 2018, the FDA established a quality assessment system called KASA (knowledge-aided assessment and structured application). The intention of KASA is to streamline the review process, make it more transparent, and meet the ever-increasing demand for faster approvals.
The system enables efficient knowledge management and establishes rules and algorithms for risk assessment of products. It enables computer-aided analyses of different aspects of the portfolio and the way they apply to existing regulations and similar products. With a harmonized structure to assess quality risks across different products, the system minimizes the need for narratives and summaries.
The KASA system is a huge step toward a digital future, but there is still a long way to go. Feeding the system with data remains human-centric, and the source documents are those that arrive at FDA’s desks, namely PDFs embedded in the eCTD. In order to fully leverage such a system, we need to complete the transition from the electronic CTD to a digital CTD.
Follow The Regulators’ Lead
In the industry, we are accustomed to bringing cutting-edge technology onto the doorsteps of the regulators and hoping they approve. As we can see, the situation with digital transformation is different and it is now the regulators leading the way. Many companies are still dragging their feet, reluctant to be first adopters of practices that appear as nice-to-haves. But as we all see in recent technological advancements, data sciences move much faster than pharmaceutical development, and the initiatives mentioned above will probably mature very quickly, which could well mean they become a regulatory standard in the very near future. It is prudent to start thinking of your own projects and start following the steps of FAIR-ification to identify the atoms of our data, and even following FDA’s proposed structures.
Another important step is harmonizing the technical terminology, which must be very precise and well defined to enable the transfer into structured systems.
The end game is to populate some form of a knowledge management system. It could be an ad-hoc company database, an existing system that is creatively adjusted, or an off-the-shelf solution like QbDVision and others.
Either way, these actions take time and require a learning curve, therefore:
- start small: one project/aspect at a time
- assign a leader to the task, and
- include digital transformation in the long-term goals of the company.
With small successes, the appetite will grow and soon enough, the team will not understand how this was ever done with pen and paper.
The Age Of The Machines
Times are changing at ever-increasing speeds, and we see the disruption the world is undergoing with the utilization of machine learning and AI. The foundations of digital transformation we laid here and the relatively modest improvements we discussed in the regulatory process are just the tip of the iceberg when it comes to the full potential of harnessing the Big Data we produce in pharmaceutical development as a whole, and in CMC particularly.
In another recent call, FDA is asking the industry to participate in a mutual reflection on the powers of AI/ML and how they will transform every aspect of the process. The prospects they list there are jaw-dropping and make the case for digital transformation even more urgent.
Just as we remember the days of trucks full of paper submissions and shake our heads in disbelief, we will probably look back to these days of eCTDs, PDFs, and Excel sheets with a similar sense of awe at the level of achievement we were able to attain before stepping aside and letting machines do the heavy lifting for us.
With the augmented abilities unlocked by machines, humans will be free to deal with the slower, ambiguous processes inherent to drug development, such as reasoning, ethical considerations, and creativity. The combination of the machines’ superpowers with human ingenuity will enable cutting-edge technologies to reach the market at faster speeds and lower costs, and increase the reliability and robustness of the drug development process as a whole.
About The Author:
 Raz Eliav is the founder of Beyond CMC, which is a consultancy that helps startups in drug development leverage the existing knowledge, know-how, and data technologies in reducing development risks, with a focus on the drug quality and manufacturing aspect, known as CMC. Eliav offers strategic consulting in CMC development, operations, and regulatory affairs, and training courses in those realms. He brings over 12 years of experience in all clinical development phases and diverse product modalities, mainly biologicals and advanced therapies.
Raz Eliav is the founder of Beyond CMC, which is a consultancy that helps startups in drug development leverage the existing knowledge, know-how, and data technologies in reducing development risks, with a focus on the drug quality and manufacturing aspect, known as CMC. Eliav offers strategic consulting in CMC development, operations, and regulatory affairs, and training courses in those realms. He brings over 12 years of experience in all clinical development phases and diverse product modalities, mainly biologicals and advanced therapies.