
Biosimilars Pipeline Shows Remarkable, Sustained Growth
By Ronald A. Rader, BioPlan Associates, Inc.
The biosimilars pipeline is progressing rapidly and continues to grow, with more products and more organizations involved. This article reviews the current biosimilars development pipeline and updates the industry’s progress.1 Quite a lot has changed in just one and a half years, and it still is early in the evolution of biosimilars, with more progress and changes coming.
involved. This article reviews the current biosimilars development pipeline and updates the industry’s progress.1 Quite a lot has changed in just one and a half years, and it still is early in the evolution of biosimilars, with more progress and changes coming.
Terminology
Regarding terminology in this article, “biosimilars” is used broadly and includes both “genuine” biosimilars, the type most familiar to those in relatively affluent developed countries, and “biogenerics,” the subset mostly distributed in poorer developing countries.2 “Genuine” biosimilars are primarily initially targeted to highly regulated, developed country markets. These undergo extensive analytical and clinical testing — particularly looking at pharmacokinetics compared to the biosimilar’s reference product, the established product it seeks to copy or emulate — and are manufactured at GMP standards
“Biogenerics” are often referred to by developers and others as biosimilars. Biogenerics similarly involve copies of reference products but are generally initially targeted to less- and nonregulated countries and international commerce. These products do not receive extensive comparative analytical and clinical testing vs. their reference product and are not manufactured to GMP standards. In the extreme, all that is needed for some countries to allow domestic distribution of biogenerics, often purchased by governments through tenders (RFPs) for supply contracts, is for the biosimilar products to meet published pharmacopeial standards.
“Follow-on” products include all reference product-derivative or me-too product classes — biosimilars, biogenerics, and biobetters. “Biobetters” are another class of follow-on biopharmaceuticals for which some data is presented in this article. These involve products that include the reference product’s active agent in some altered form, whether it has been molecularly modified or, often it is a substructure, such as a pegylated active agent. In some cases, the finished product is different, such as those containing the same active agent in an orally administered vs. injectable format.
Pipeline Growth
The growth in the biopharmaceuticals follow-on product development pipeline is shown in Table 1. Data is derived from the Biosimilars/Biobetters Pipeline Directory and represents the number of relevant entries in the database at that time.3 Biosimilars in the pipeline are still robust, having grown at an average of greater than 20 percent annually in the past one and a half years. This sustained growth is remarkable, given that over the past five and a half years the pipeline has expanded at an average of 27 percent annually.
Growth in the number of approved products is expected to continue, due to more biosimilars entering development, including a wave of reference products in the U.S. coming off patent starting this year. At some point, the biosimilar (and their competing products) market will become too crowded, but that will likely be years off. Competition among biosimilars and their reference products is essentially just getting started in most markets, including the U.S., while there is more competition in the longer-established Western European markets.
Table 1: Number of Follow-on Products in the Pipeline, Annual Growth Rates
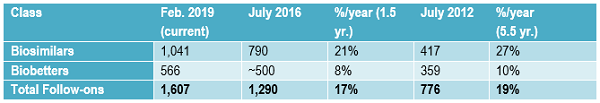
Compared to biosimilars, the rate of biobetters development has generally remained rather slow and steady over time. Biobetters are a class of partially innovative “me-too” versions of biopharmaceutical products. They are not new at all (e.g., recombinant insulin is a biobetter version of animal-derived insulin) compared to biosimilars, which are a new regulatory approvals-defined class of products. Biobetters involve changes in products, generally involving something novel. They need to receive full, original approvals, e.g., full BLAs, with clinical development including costly full traditional preclinical testing through Phase 3 placebo-controlled clinical trials.
Biologics Vs. Drugs At The FDA
FDA regulatory classifications of some older-type biopharmaceutical products are now in the process of being shifted from being treated as drugs (receiving NDA approvals) to biologics (receiving BLA approvals), including biosimilar approvals. This includes five products (see Table 2) receiving biosimilar-like approvals as generic drugs, 505(b)(2)-type NDA approvals, with the reference products still regulated as drugs. These products include various human- and animal-derived products, such as porcine and bovine insulin and somatropin (human growth hormone). These have traditionally been regulated as drugs and have been replaced by recombinant (biobetter) versions. These products are now transitioning to being classified as biologics, with their approvals including biosimilar approvals, with existing approvals being converted from NDAs to BLAs.
Despite drugs vs. biologics regulations being better aligned, regulation of biosimilars and broader biopharmaceuticals by the FDA remains divided between two entities — the Center for Biologics Evaluation and Research (CBER) and the Center for Drugs Evaluation and Research (CDER). Contrary to most expectations, CDER, the “drugs” center, now approves and regulates the majority of biopharmaceuticals, particularly those that are relatively readily characterizable or considered single active agent products, including recombinant proteins and antibodies. CBER continues to handle traditionally more complex biological and living organism-derived products, including vaccines, blood/plasma-derived products, and cellular and gene therapies. However, the great majority of biosimilars and biopharmaceuticals continue to be recombinant proteins or antibodies handled by CDER.
To date, all biosimilar approvals have involved recombinant proteins/antibodies and have been granted by CDER. This is despite the fact that all products regulated as biologics, including the CBER-regulated therapeutics, are fully capable of receiving biosimilar approvals. However, we may never see biosimilar approvals of the more biological-type products CBER regulates, since the great majority of these products essentially have and continue to receive full BLA approvals that are much like biosimilars, with the approvals primarily based on comparative clinical trials and (bio)analytical studies. For example, unless involving a new antigen, the great majority of new vaccines (targeting the same diseases), such as new influenza or pertussis vaccines, or blood-derived products, such as factor VIII or albumin, receive approvals based on trials and (bio)analytical studies comparing them with a prior established (reference) vaccine product. Such full approvals, besides supporting better brand image for that product than biosimilar approval, provide 12 years of exclusivity protection for that reference product from biosimilar approval.
FDA Approvals
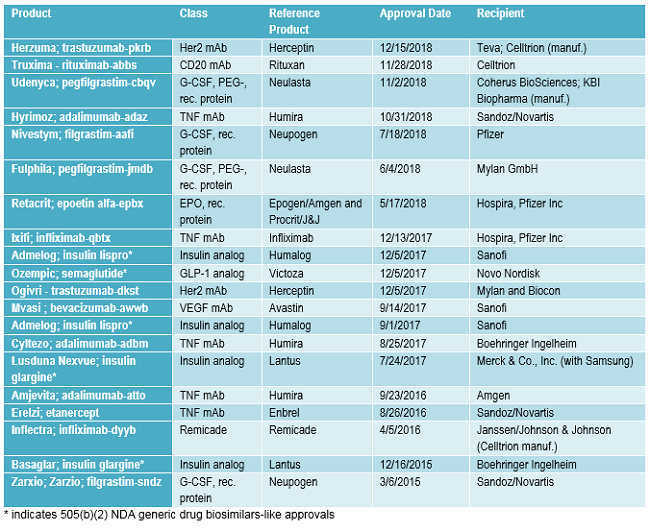
FDA approvals of biosimilars and very closely related 505(b)(2) generic drug approvals granted to biopharmaceuticals being transitioned to biosimilars approvals to date are listed in Table 2. Following the pattern discussed below, with multiple biosimilars targeting the same successful reference products, 13 (65 percent) of the 20 approvals listed involve the same reference products, e.g., there are multiple biosimilar versions of Humira. Also, note that many of the same companies are involved, too.
Table 2: Biosimilar BLA Approvals and Rather Similar 505(b)(2) Biopharmaceutical Approvals Since the First Biosimilar Approval
Popular Biosimilar Targets
For every potentially commercially viable reference product with approaching patent expirations or lacking protection (e.g., in developing countries or expired), there are multiple biosimilars in the pipeline.1 Table 3 shows the growth in the number of biosimilars in the pipeline for some of the most “popular” reference product targets now and approximately one and a half years ago. The number of biosimilars in the pipeline, most still in development, targeting the same popular reference products has increased in the past 18 months at a rate averaging over 50 percent annually. This very high growth rate in the biosimilars pipeline is reflected in the growth in the numbers of biosimilars targeting other less-popular reference product targets, too.
Table 3: Number of Biosimilars in the Development Pipeline for Some Major Reference Product and Classes Now and ~1.5 Years Ago
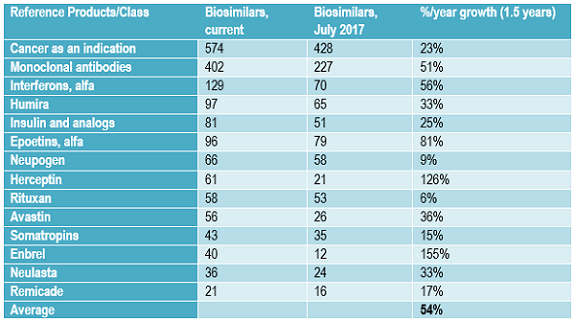
This growth in number of biosimilars in development targeting the same reference products will likely continue in the short term, through the next few years, as more current biosimilar players extend their portfolios of products in development and new developers continue to enter the field. Many biosimilar developers, particularly those planning to market these products themselves, plan to eventually have a portfolio of multiple biosimilars, whether as a distinct line of products or to add to existing disease-oriented product portfolios. With most biosimilars in the pipeline from companies currently lacking significant marketing presence in major markets, most of these products are expected to be licensed-in for marketing by companies seeking to develop broader portfolios of biosimilar products, often with full rights, including manufacturing.
Biosimilars Names
Since July 2016, the FDA has finalized and adopted certain rules for biosimilar nomenclature. These rules involve “proper” names, the arbitrary (made-up) non-proprietary (no restrictions on use) short names used in labeling, designating prescriptions, etc., generally with 1:1 association with the trademark used. Essentially, the FDA adopted the International Nonproprietary Name (INN; which is generally adopted as the U.S. Adopted Name/USAN), an inherently generic name assigned by the World Health Organization (WHO)/UN to products with closely matching active agent structures or compositions (e.g., epoetin alfa is assigned to human erythropoietin (EPO) products), and then a random four-letter suffix is appended to uniquely identify specific approved products. For example, infliximab-dyyb is the proper name assigned to the product with the Inflectra trademark, with “infliximab” alone applying to multiple products.
But problems with biosimilar (and biopharmaceutical) nomenclature persist and will worsen as time goes on and the number of regulated products worldwide increases. This includes proper names being approvals-centric, assigned to products as defined in their approved applications and not based on product identity or conveying much information about the product. The FDA and other regulators have avoided nomenclature system (rules)-based unique names for biologics. This includes the FDA having no active agent or finished product nomenclature providing unique names or identifiers based on agent/product identity (structure, composition, etc.). With no unique names for active agents, this results in the same product’s proper or active agent generic name being used for both the official product name and the product’s active agent. Despite this potentially affecting patient safety, the FDA is showing no indication of working on nomenclature systems that will uniquely identify biosimilar (and other biopharmaceutical) active agents.
The problems with the FDA’s proper names for biologics mostly result from the adopted rules being a compromise capping decade-long public controversies between interested parties seeking to have proper names be fully unique rather than more generic names/identifiers for biosimilar products. The resulting proper names are still not fully optimal as unique or generic identifiers. For example, healthcare professionals need to know or look up products’ proper names, particularly their unique random four-letter suffixes, and the use of the same generic active agent name as the root name for products sharing biosimilarity can contribute to mix-ups among these similar products. Also, proper names cover only biologic products upon approval, which currently will not change, even with “product drift,” i.e., manufacturing processes, specifications, formulation, and/or other changes in products granted through supplemental approvals (with reference and other product drift simply not yet being reported by the FDA).
Conclusion
Biosimilars involve many new products, players, and manufacturing facilities joining the world’s biopharmaceutical industry. The biosimilars development pipeline and number of product approvals continue to grow. This includes many developers targeting biosimilars of many of the same reference products, and competition among biosimilars (including their reference products) will increase significantly in coming years. Product regulation continues to shift, such as, for example, approvals being shifted from “drugs” to “biologics,” and biosimilar naming/identifier issues remain only partially resolved.
Participate in BioPlan Associates’ 16th Annual Biopharmaceutical Manufacturing Report and Survey, the bioindustry's most in-depth, global benchmarking and analysis study. Participants will receive an aggregated summary of the survey results along with the white paper Top Trends in Biopharmaceutical Manufacturing. Click here to participate in the survey.
References:
- Rader, R.A.,"Biosimilars Pipeline Analysis: Many Products, More Competition Coming", Biosimilar Development, July 26, 2016, https://www.bioprocessonline.com/doc/biosimilars-pipeline-analysis-many-products-more-competition-coming-0001
- Rader, R.A., "Biosimilars in the Rest of the World: Developments in Lesser-Regulated Countries," BioProcessing J., 12(4), Winter 2013/2014, p. 41-47
- Rader, R.A., Biosimilars/Biobetters Pipeline Directory, online database (www.biosimilarspipeline.com) and derived PDF report.
About The Author:
 Ronald A. Rader is senior director of technical research at BioPlan Associates. He has 35+ years’ experience as a biotechnology and pharmaceutical — particularly biopharmaceutical — information specialist, analyst, and publisher. Publications/information resources has been responsible for include the Antiviral Agents Bulletin periodical, the Federal Bio-Technology Transfer Directory, BIOPHARMA: Biopharmaceutical Products in the U.S. and European Markets, and the Biosimilars/Biobetters Pipeline Directory. He can be reached at info@bioplanassociates.com or (301) 921-5979.
Ronald A. Rader is senior director of technical research at BioPlan Associates. He has 35+ years’ experience as a biotechnology and pharmaceutical — particularly biopharmaceutical — information specialist, analyst, and publisher. Publications/information resources has been responsible for include the Antiviral Agents Bulletin periodical, the Federal Bio-Technology Transfer Directory, BIOPHARMA: Biopharmaceutical Products in the U.S. and European Markets, and the Biosimilars/Biobetters Pipeline Directory. He can be reached at info@bioplanassociates.com or (301) 921-5979.